1 Name of Medicine
Dostarlimab.
2 Qualitative and Quantitative Composition
Each mL of concentrate for solution for infusion contains 50 mg of dostarlimab.
One vial of 10 mL concentrate for solution for infusion contains 500 mg of dostarlimab (50 mg/mL).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrate for solution for infusion (sterile concentrate).
Dostarlimab is a clear to slightly opalescent colourless to yellow solution, free from visible particles.
4.1 Therapeutic Indications
Jemperli is indicated in combination with carboplatin and paclitaxel for the treatment of adult patients with primary advanced or recurrent endometrial cancer.
Jemperli is indicated as monotherapy for the treatment of adult patients with recurrent or advanced mismatch repair deficient (dMMR) endometrial cancer (EC) that has progressed on or following prior treatment with a platinum-containing regimen.
4.2 Dose and Method of Administration
Dostarlimab in combination with chemotherapy.
When dostarlimab is administered in combination with chemotherapy, refer to the full Product Information for the combination products (also see Section 5.1 Pharmacodynamic Properties, Clinical trials).
The recommended dose as combination therapy is 500 mg dostarlimab administered as an intravenous infusion over 30 minutes every 3 weeks for 6 doses followed by 1000 mg every 6 weeks for all cycles thereafter.
The dosage regimen in combination with chemotherapy is presented in Table 1.
 Administration of dostarlimab should continue according to the recommended dose and schedule until disease progression or unacceptable toxicity.
Administration of dostarlimab should continue according to the recommended dose and schedule until disease progression or unacceptable toxicity.
Dostarlimab monotherapy.
The recommended dose as monotherapy is 500 mg dostarlimab administered as an intravenous infusion over 30 minutes every 3 weeks for 4 doses followed by 1,000 mg every 6 weeks for all cycles thereafter.
The dosage regimen as a monotherapy is presented in Table 2.
 Administration of dostarlimab should continue according to the recommended dose and schedule until disease progression or unacceptable toxicity.
Administration of dostarlimab should continue according to the recommended dose and schedule until disease progression or unacceptable toxicity.
Dose modifications.
Dose reduction is not recommended. Dosing delay or discontinuation may be required based on individual safety and tolerability. Recommended modifications to manage adverse reactions are provided in Table 3. Detailed guidelines for the management of immune-related adverse reactions and infusion-related reactions are described in Section 4.4 Special Warnings and Precautions for Use.

Method of administration.
Preparation.
Parenteral medicinal products should be inspected visually for particulate matter and discolouration prior to administration. Dostarlimab is a slightly opalescent colourless to yellow solution. Discard the vial if visible particles are observed.
Dilution.
For the 500-mg dose, withdraw 10 mL of dostarlimab from a vial and transfer into an intravenous (IV) bag containing sodium chloride 9 mg/mL (0.9%) solution for injection, or glucose 50 mg/mL (5%) solution for injection. The final concentration of the diluted solution should be between 2 mg/mL and 10 mg/mL. The total volume of the infusion solution must not exceed 250 mL. This may require withdrawing a volume of diluent from the IV bag prior to adding a volume of dostarlimab into the IV bag.
For example, if preparing a 500 mg dose in a 250 mL diluent IV bag, to achieve a 2 mg/mL concentration would require withdrawing 10 mL of diluent from the 250 mL IV bag. Then, 10 mL of dostarlimab would be withdrawn from the vial and transferred into the IV bag.
For the 1,000-mg dose, withdraw 10 mL of dostarlimab from each of two vials (withdraw 20 mL total) and transfer into an IV bag containing sodium chloride 9 mg/mL (0.9%) solution for injection or glucose 50 mg/mL (5%) solution for injection. The final concentration of the diluted solution should be between 4 mg/mL and 10 mg/mL. The total volume of the infusion solution must not exceed 250 mL. This may require withdrawing a volume of diluent from the IV bag prior to adding a volume of dostarlimab into the IV bag.
For example, if preparing a 1,000 mg dose in a 250 mL diluent IV bag, to achieve a 4 mg/mL concentration would require withdrawing 20 mL of diluent from the 250 mL IV bag. Then, 10 mL of dostarlimab would be withdrawn from each of two vials, totaling 20 mL, and transferred into the IV bag.
Mix diluted solution by gentle inversion. Do not shake the final infusion bag. Discard any unused portion left in the vial.
Administration.
Dostarlimab is for intravenous infusion only. Dostarlimab should be administered by intravenous infusion using an intravenous infusion pump over 30 minutes by a health care practitioner.
Dostarlimab must not be administered as an intravenous push or bolus injection.
Dostarlimab is compatible with an IV bag made of polyvinyl chloride (PVC) with or without di(2-ethylhexyl) phthalate (DEHP), ethylene vinyl acetate, polyethylene (PE), polypropylene (PP) or polyolefin blend (PP+PE), and a syringe made from PP. Infusion tubing should be made of PVC, platinum cured silicon or PP; fittings made from PVC or polycarbonate and needles made from stainless steel. A 0.2 or 0.22 micron in-line polyethersulfone (PES) filter must be used during administration of dostarlimab.
Children.
The safety and efficacy of dostarlimab in children and adolescents aged under 18 years have not been established. No data are available.
Elderly.
No dose adjustment is recommended for patients who are 65 years of age or over. There are limited clinical data with dostarlimab in patients 75 years of age or over (see Section 5.1 Pharmacodynamic Properties).
Renal impairment.
No dose adjustment is recommended for patients with mild or moderate renal impairment. There are limited data in patients with severe renal impairment or end-stage renal disease undergoing dialysis (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
No dose adjustment is recommended for patients with mild hepatic impairment. There are limited data in patients with moderate or severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
Jemperli is contraindicated in patients with a known hypersensitivity to dostarlimab or any of the excipients.
4.4 Special Warnings and Precautions for Use
Traceability.
In order to improve the traceability of biological medicinal products, the tradename and the batch number of the administered product should be clearly recorded.
Immune-related adverse reactions.
Immune-related adverse reactions, which may be severe or fatal, can occur in patients treated with antibodies blocking the programmed cell death protein-1/ programmed death-ligand 1 (PD-1/PD-L1) pathway, including dostarlimab. While immune-related adverse reactions usually occur during treatment with PD-1/PD-L1 blocking antibodies, symptoms can also manifest after discontinuation of treatment. Immune-related adverse reactions may occur in any organ or tissue and may affect more than one body system simultaneously. Important immune-related adverse reactions listed in this section are not inclusive of all possible severe and fatal immune-related reactions.
Early identification and management of immune-related adverse reactions are essential to ensure safe use of PD-1/PD-L1 blocking antibodies. Monitor for symptoms and signs of immune-related adverse reactions. Evaluate haematological and clinical chemistries, including liver, kidney and thyroid function tests, at baseline and periodically during treatment. For suspected immune-related adverse reactions, adequate evaluation including specialty consultation should be ensured.
Based on the severity of the adverse reaction, dostarlimab should be withheld or permanently discontinued and corticosteroids (1 to 2 mg/kg/day prednisone or equivalent) or other appropriate therapy administered (see below and see Section 4.2 Dose and Method of Administration, Dose modifications). Upon improvement to Grade 0 or 1, corticosteroid taper should be initiated and continued for 1 month or longer. Based on limited data from clinical studies in patients whose immune-related adverse reactions could not be controlled with corticosteroid use, administration of other systemic immunosuppressants can be considered. Institute hormone replacement therapy for endocrinopathies as warranted.
Dostarlimab should be permanently discontinued for any Grade 3 immune-related adverse reaction that recurs and for any Grade 4 immune-related adverse reaction toxicity, except for endocrinopathies that are controlled with replacement hormones and unless otherwise specified in Table 3.
Immune-related pneumonitis. Pneumonitis has been reported in patients receiving dostarlimab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of pneumonitis. Suspected pneumonitis should be confirmed with radiographic imaging and other causes excluded. Patients should be managed with dostarlimab treatment modifications and corticosteroids (see Section 4.2 Dose and Method of Administration).
Immune-related colitis. Dostarlimab can cause immune-related colitis (see Section 4.8 Adverse Effects (Undesirable Effects)). Monitor patients for signs and symptoms of colitis and manage with dostarlimab treatment modifications, anti-diarrhoeal agents and corticosteroids (see Section 4.2 Dose and Method of Administration).
Immune-related hepatitis. Dostarlimab can cause immune-related hepatitis. Monitor patients for changes in liver function periodically as indicated based on clinical evaluation and manage with dostarlimab treatment modifications and corticosteroids (see Section 4.2 Dose and Method of Administration).
Immune-related endocrinopathies. Immune-related endocrinopathies, including hypothyroidism, hyperthyroidism, thyroiditis, hypophysitis, type 1 diabetes mellitus, diabetic ketoacidosis and adrenal insufficiency, have been reported in patients receiving dostarlimab (see Section 4.8 Adverse Effects (Undesirable Effects)).
Hypothyroidism and hyperthyroidism.
Immune-related hypothyroidism and hyperthyroidism (including thyroiditis) occurred in patients receiving dostarlimab, and hypothyroidism may follow hyperthyroidism. Patients should be monitored for abnormal thyroid function tests prior to and periodically during treatment and as indicated based on clinical evaluation. Immune-related hypothyroidism and hyperthyroidism (including thyroiditis) should be managed as recommended in Section 4.2 Dose and Method of Administration.
Adrenal insufficiency.
Immune-related adrenal insufficiency occurred in patients receiving dostarlimab. Patients should be monitored for clinical signs and symptoms of adrenal insufficiency. For symptomatic adrenal insufficiency, patients should be managed as recommended in Section 4.2 Dose and Method of Administration.
Immune-related nephritis. Dostarlimab can cause immune-related nephritis (see Section 4.8 Adverse Effects (Undesirable Effects)). Monitor patients for changes in renal function and manage with dostarlimab treatment modifications and corticosteroids (see Section 4.2 Dose and Method of Administration).
Immune related rash. Immune-related rash has been reported in patients receiving dostarlimab, including pemphigoid (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of rash. Exfoliative dermatologic conditions should be managed as recommended (see Section 4.2 Dose and Method of Administration). Events of Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN) or drug rash with eosinophilia and systemic symptoms (DRESS) have been reported in patients treated with PD-1 inhibitors.
Caution should be used when considering the use of dostarlimab in a patient who has previously experienced a severe or life-threatening skin adverse reaction on prior treatment with other immune-stimulatory anticancer agents.
Other immune-related adverse reactions. Given the mechanism of action of dostarlimab other potential immune-related adverse reactions may occur. Clinically significant immune-related adverse reactions reported in less than 1% of patients treated with dostarlimab in clinical trials or observed during treatment with other immune checkpoint inhibitors include Guillain-Barre syndrome, encephalitis, autoimmune haemolytic anaemia, aplastic anaemia, haemophagocytic lymphohistiocytosis (HLH), uveitis, and iridocyclitis. Patients should be monitored for signs and symptoms of immune-related adverse reactions and managed as described in Section 4.2 Dose and Method of Administration.
Patients with pre-existing autoimmune disease (AID).
In patients with pre-existing autoimmune disease (AID), data from observational studies suggest that the risk of immune-mediated adverse reactions following immune checkpoint inhibitor therapy may be increased as compared with the risk in patients without pre-existing AID. In addition, flares of the underlying AID were frequent, but the majority were mild and manageable.
Transplant-related adverse reactions.
Solid organ transplant rejection.
Solid organ transplant rejection has been reported in the postmarketing setting in patients treated with PD-1 inhibitors. Treatment with dostarlimab may increase the risk of rejection in solid organ transplant recipients. The benefit of treatment with dostarlimab versus the risk of possible organ rejection should be considered in these patients.
Complications of allogeneic haematopoietic stem cell transplant (HSCT).
Fatal and other serious complications can occur in patients who receive allogeneic haematopoietic stem cell transplantation (HSCT) before or after being treated with a PD-1/PD-L1-blocking antibody. Transplant-related complications include hyperacute graft-versus-host disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause). These complications may occur despite intervening therapy between PD-1/PD-L1 blockade and allogeneic HSCT. Follow patients closely for evidence of transplant-related complications and intervene promptly. Consider the benefit versus risks of treatment with a PD-1/PD-L1-blocking antibody prior to or after an allogeneic HSCT.
Infusion-related reactions.
Dostarlimab can cause infusion-related reactions, which can be severe (see Section 4.8 Adverse Effects (Undesirable Effects)). For severe (Grade 3) or life-threatening (Grade 4) infusion-related reactions, stop infusion and permanently discontinue dostarlimab (see Section 4.2 Dose and Method of Administration).
Patient card.
All prescribers of Jemperli should inform patients about the patient card, explaining what to do should they experience any symptom of immune-related adverse reactions. The physician will provide the patient card to each patient.
Use in hepatic impairment.
There are limited data in patients with severe or moderate hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Use in renal impairment.
There are limited data in patients with severe renal impairment or end-stage renal disease undergoing dialysis (see Section 5.2 Pharmacokinetic Properties).
Use in the elderly.
There are limited clinical data with dostarlimab in patients aged 75 years or over (see Section 5.1 Pharmacodynamic Properties).
Paediatric use.
No data are available.
Effects on laboratory tests.
No data are available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No drug-drug interaction studies have been conducted with dostarlimab. Monoclonal antibodies (mAbs) such as dostarlimab are not substrates for cytochrome P450 or drug transporters. Additionally, pharmacokinetic (PK) drug-drug interaction of dostarlimab with small molecule drugs is not expected.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Fertility studies have not been conducted with dostarlimab. No effects on male and female reproductive organs were observed in a 3-month repeat dose toxicity study in cynomolgus monkeys at ≤ 100 mg/kg/week IV, resulting in exposures (AUC) at least 28 times that expected in patients; however, these results may not be predictive of clinical risk because of the immaturity of the reproductive system of animals used in the study.
(Category D)
There are no available data on the use of dostarlimab in pregnant women. Animal reproduction studies have not been conducted with dostarlimab to evaluate its effect on reproduction and fetal development. Based on its mechanism of action, dostarlimab can cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PD-L1 signalling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. Human IgG4 immunoglobulins (IgG4) are known to cross the placental barrier; therefore, dostarlimab has the potential to be transmitted from the mother to the developing fetus. Advise women of the potential risk to a fetus.
Dostarlimab is not recommended during pregnancy. Women of childbearing potential should use highly effective contraception during treatment with dostarlimab and for 4 months after the last dose.
There is no information regarding the presence of dostarlimab in human milk, or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment and for 4 months after the last dose of dostarlimab.4.7 Effects on Ability to Drive and Use Machines
Dostarlimab has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Clinical trial data.
Summary of the safety profile. Dostarlimab in combination with carboplatin and paclitaxel.
The safety of dostarlimab in combination with chemotherapy has been evaluated in 241 patients with primary advanced or recurrent EC in the RUBY study. Patients received doses of dostarlimab 500 mg every 3 weeks for 6 cycles followed by 1000 mg every 6 weeks for all cycles thereafter.
Serious adverse reactions occurred in 26% of patients receiving dostarlimab in combination with carboplatin and paclitaxel; the most common serious adverse reaction was sepsis, including urosepsis (3.7%). Fatal adverse reactions occurred in 1.2% of patients receiving dostarlimab including septic shock (0.8%), and myelosuppression (0.4%).
In patients receiving dostarlimab in combination with carboplatin and paclitaxel, dostarlimab was permanently discontinued due to adverse reactions in 24 patients (10%). Adverse reactions that required permanent discontinuation in ≥ 2 patients included 3 cases (1.2%) of rash maculo-papular, and 2 cases (0.8%) each of alanine aminotransferase increased, aspartate aminotransferase increased, diarrhea, pancreatitis, fatigue, pneumonitis and arthralgia.
Dosage interruptions due to an adverse reaction occurred in 37% of patients who received dostarlimab in combination with carboplatin and paclitaxel. Adverse reactions that required dosage interruption in ≥ 5% of patients who received dostarlimab in combination with carboplatin and paclitaxel were anaemia, thrombocytopenia and peripheral neuropathy.
The most common adverse reactions, including laboratory abnormalities (≥ 20%), were decreased haemoglobin, decreased white blood cell count, decreased lymphocytes, increased glucose, decreased neutrophils, increased aspartate aminotransferase, rash, increased alanine aminotransferase and increased alkaline phosphatase.
Adverse reactions observed in EC patients are presented in Table 4.
Adverse reactions known to occur with dostarlimab or with combination therapy components given alone may occur during treatment with these medicinal products in combination, even if these reactions were not reported in clinical studies with combination therapy.
When dostarlimab is administered in combination, refer to the local Product Information for the respective combination therapy component prior to initiation of treatment.
Adverse reactions are presented by system organ class and by frequency. Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); and not known (cannot be estimated from the available data).
 Table 5 summarises other adverse events that occurred in 10% or more of patients with endometrial cancer treated with dostarlimab in combination with chemotherapy in the RUBY study.
Table 5 summarises other adverse events that occurred in 10% or more of patients with endometrial cancer treated with dostarlimab in combination with chemotherapy in the RUBY study.

Dostarlimab as monotherapy.
The safety of dostarlimab as monotherapy has been evaluated in 605 patients with recurrent or advanced solid tumours including 314 patients with endometrial cancer and 291 patients with other advanced solid tumours, in the GARNET study. Patients received doses of dostarlimab 500 mg every 3 weeks for 4 cycles followed by 1000 mg every 6 weeks for all cycles thereafter.
Adverse reactions observed in patients who received dostarlimab monotherapy in the open-label, multicohort GARNET study are listed in Table 6.
Adverse reactions are presented by system organ class and by frequency. Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); and not known (cannot be estimated from the available data).
 Other adverse events. Table 7 summarises other adverse events that occurred in 10% or more of patients with solid tumours treated with dostarlimab monotherapy in the GARNET study. Fatigue and asthenia are the only adverse events reported in at least 20% of patients. Grade 4 events included asthenia (N=1 patient, 0.2%) and dyspnoea (N=2 patients, 0.3%).
Other adverse events. Table 7 summarises other adverse events that occurred in 10% or more of patients with solid tumours treated with dostarlimab monotherapy in the GARNET study. Fatigue and asthenia are the only adverse events reported in at least 20% of patients. Grade 4 events included asthenia (N=1 patient, 0.2%) and dyspnoea (N=2 patients, 0.3%).

Immunogenicity.
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralising antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to dostarlimab in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In the GARNET study, anti-drug antibodies (ADA) were tested in 384 patients who received dostarlimab monotherapy and the incidence of dostarlimab treatment-emergent ADAs was 2.1%. Neutralising antibodies were detected in 1.0% of patients.
Co-administration with chemotherapy did not affect dostarlimab immunogenicity. In the RUBY study, of the 225 patients who were treated with dostarlimab in combination with chemotherapy and evaluable for the presence of ADAs, there was no incidence of dostarlimab treatment-emergent ADA or treatment emergent neutralising antibodies.
In the patients who developed anti-dostarlimab antibodies in either study, there was no evidence of altered pharmacokinetics, efficacy or safety of dostarlimab. Because of the small number of patients who developed ADAs, the impact of immunogenicity on the efficacy and safety of dostarlimab is inconclusive.
Immune checkpoint inhibitor class effects.
There have been cases of the following adverse reactions reported during treatment with other immune checkpoint inhibitors which might also occur during treatment with dostarlimab: coeliac disease; pancreatic exocrine insufficiency.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
If overdose is suspected, the patient should be monitored for any signs or symptoms of adverse reactions or effects, and appropriate standard of care measures should be instituted immediately.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Anti-neoplastic agents, monoclonal antibodies.
ATC code: L01FF07.
Mechanism of action.
Dostarlimab is an anti-programmed cell death protein-1 (PD-1) immunoglobulin G4 (IgG4) humanised monoclonal antibody (mAb), derived from a stable Chinese hamster ovary (CHO) cell line.
Binding of the PD-1 ligands, PD-L1 and PD-L2, to the PD-1 receptor found on T cells inhibits T-cell proliferation and cytokine production. Upregulation of PD-1 ligands occurs in some tumours and signalling through this pathway can contribute to inhibition of active T-cell immune surveillance of tumours. Dostarlimab is a humanised mAb of IgG4 isotype that binds to PD 1, resulting in inhibition of binding to PD-L1 and PD-L2, releasing inhibition of PD-1 pathway-mediated immune response, including the anti-tumour immune response. In syngeneic mouse tumour models, blocking PD-1 activity resulted in decreased tumour growth.
Pharmacodynamic effects.
Based on exposure efficacy and safety relationships, there are no clinically significant differences in efficacy and safety within the exposure range attained under the recommended therapeutic dosing regimen (500 mg administered intravenously every 3 weeks for 4 doses, followed by 1,000 mg every 6 weeks thereafter). Full receptor occupancy as measured by both the direct PD-1 binding and IL-2 production functional assay was maintained throughout the dosing interval at the recommended therapeutic dosing regimen.
Clinical trials.
RUBY: randomised controlled study of combination therapy in treatment of primary advanced or recurrent EC.
The efficacy and safety of dostarlimab in combination with carboplatin-paclitaxel were investigated in RUBY, a multicentre, randomised, double-blinded, placebo-controlled Phase 3 study conducted in patients with primary advanced or recurrent EC.
Patients were randomised (1:1) to receive dostarlimab 500 mg plus carboplatin AUC 5 mg/mL/min and paclitaxel 175 mg/m2 every 3 weeks for 6 cycles followed by dostarlimab 1000 mg every 6 weeks (n = 245) or placebo plus carboplatin AUC 5 mg/mL/min and paclitaxel 175 mg/m2 every 3 weeks for 6 cycles followed by placebo every 6 weeks (n = 249). Randomisation was stratified by MMR/MSI status, prior external pelvic radiotherapy, and disease status (recurrent, primary Stage III, or primary Stage IV).
The key eligibility criteria for the study were International Federation of Gynaecology and Obstetrics (FIGO) primary Stage III or Stage IV disease, including Stage IIIA to IIIC1 disease with presence of evaluable or measurable disease per RECIST v.1.1, Stage IIIC1 patients with carcinosarcoma, clear cell, serous, or mixed histology (containing ≥ 10% carcinosarcoma, clear cell, or serous histology) regardless of presence of evaluable or measurable disease on imaging, Stage IIIC2 or Stage IV disease regardless of presence of evaluable or measurable disease. The study also included patients with first recurrent EC with a low potential for cure by radiation therapy or surgery alone or in combination, including patients who had first recurrent disease and were naïve to systemic anticancer therapy or who had received prior neo-adjuvant/adjuvant systemic anticancer therapy and had a recurrence or progressive disease ≥ 6 months after completing treatment (first recurrence). Treatment continued for up to 3 years or until unacceptable toxicity, disease progression or investigator decision. Assessment of tumour status was performed every 6 weeks through week 25, every 9 weeks through week 52 and every 12 weeks thereafter.
The primary efficacy outcome measures were progression-free survival (PFS), assessed by the investigator according to RECIST v1.1 in subjects with dMMR/MSI-H primary advanced or recurrent EC and in all subjects (overall population) with primary advanced or recurrent EC, and overall survival (OS) in all subjects (overall population) with primary advanced or recurrent EC. Secondary endpoints included objective response rate (ORR), duration of response (DOR), and disease control rate (DCR) as assessed by blinded independent central radiologists' (BICR) review and investigator assessment according to RECIST v1.1, and PFS2, defined as the time from treatment randomisation to the date of assessment of progression on the first subsequent anticancer therapy following study treatment or death by any cause, whichever was earlier.
A total of 494 patients with EC were evaluated for efficacy in the RUBY study. Baseline demographics and characteristics of the overall study population were: median age 65 years (51% aged 65 years or older); 77% White, 12% Black, 3% Asian; Eastern Cooperative Oncology Group (ECOG) performance score (PS) 0 (63%) or 1 (37%); primary stage III 18.6%, primary stage IV 33.6%, recurrent EC 47.8%.
The identification of dMMR/MSI-H tumour status was prospectively determined based on local testing assays (IHC, PCR or NGS), or central testing (IHC) when no local result was available.
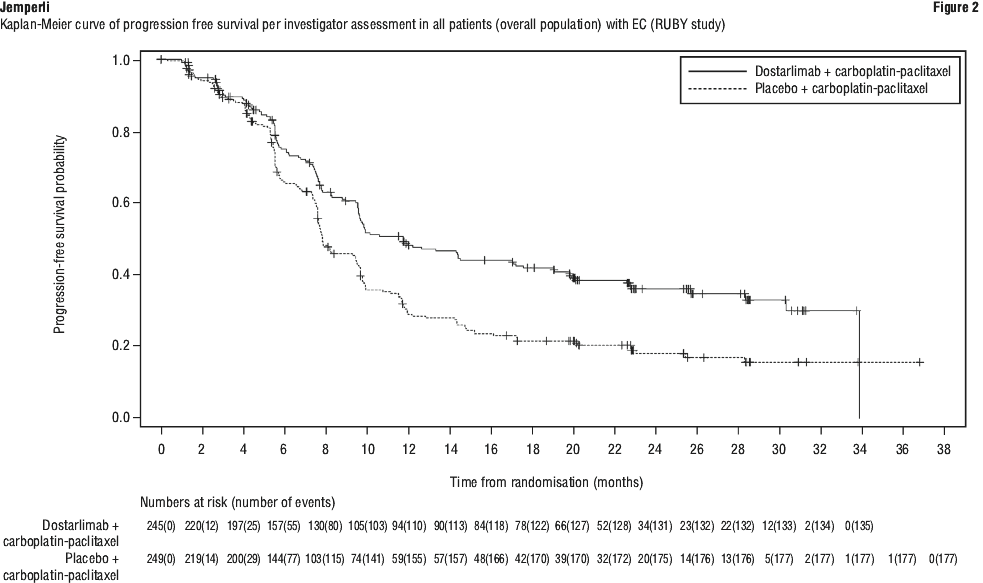
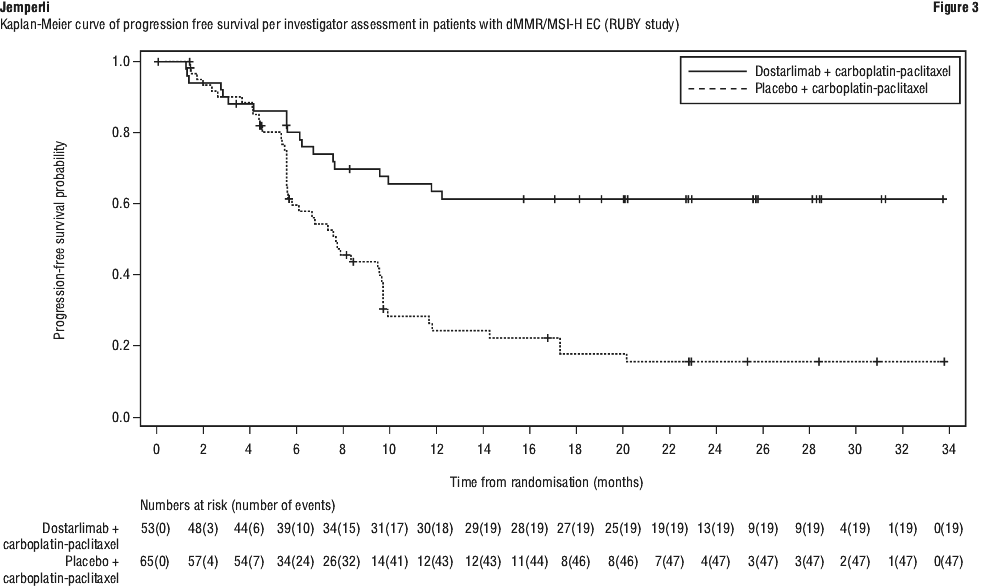
Efficacy results are shown in Table 8 and Figures 1, 2 and 3. Dostarlimab plus carboplatin-paclitaxel demonstrated statistically significant improvements in PFS in both the dMMR/MSI-H and overall populations and OS in the overall population versus placebo plus carboplatin-paclitaxel.
 Pre-specified exploratory analyses of PFS and OS were performed in patients with MMRp/MSS EC (n = 376). The PFS HR was 0.76 (95% CI: 0.59, 0.98) with a median PFS of 9.9 months for dostarlimab plus carboplatin-paclitaxel (n = 192) versus 7.9 months for placebo plus carboplatin-paclitaxel (n = 184) (cut-off date 28 Sept 2022). The OS HR was 0.79 (95% CI: 0.60, 1.04) with a median OS of 34 months for dostarlimab plus carboplatin-paclitaxel versus 27 months for placebo plus carboplatin-paclitaxel (cut-off date 22 Sept 2023).
Pre-specified exploratory analyses of PFS and OS were performed in patients with MMRp/MSS EC (n = 376). The PFS HR was 0.76 (95% CI: 0.59, 0.98) with a median PFS of 9.9 months for dostarlimab plus carboplatin-paclitaxel (n = 192) versus 7.9 months for placebo plus carboplatin-paclitaxel (n = 184) (cut-off date 28 Sept 2022). The OS HR was 0.79 (95% CI: 0.60, 1.04) with a median OS of 34 months for dostarlimab plus carboplatin-paclitaxel versus 27 months for placebo plus carboplatin-paclitaxel (cut-off date 22 Sept 2023).

 Patient-reported outcomes (PROs) were assessed using European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire (EORTC) QLQ-C30. Throughout the first 6 dosing cycles of the study, there were no clinically meaningful differences between dostarlimab plus carboplatin-paclitaxel and placebo plus carboplatin-paclitaxel arms in patient-reported symptoms, functioning, and health-related quality of life (assessed by a difference of ≥ 10 points from the baseline assessment).
Patient-reported outcomes (PROs) were assessed using European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire (EORTC) QLQ-C30. Throughout the first 6 dosing cycles of the study, there were no clinically meaningful differences between dostarlimab plus carboplatin-paclitaxel and placebo plus carboplatin-paclitaxel arms in patient-reported symptoms, functioning, and health-related quality of life (assessed by a difference of ≥ 10 points from the baseline assessment).
GARNET: patients with recurrent or advanced dMMR/MSI-H EC who have progressed on or after treatment with a platinum-containing regimen.
The efficacy and safety of dostarlimab as a monotherapy were investigated in GARNET, a multicentre, open-label, Phase 1 dose escalation study conducted in patients with recurrent or advanced EC that has progressed on or after treatment with a platinum-containing regimen.
The GARNET study included expansion cohorts in subjects with recurrent or advanced solid tumours who have limited available treatment options. Cohort A1 enrolled patients with mismatch repair deficient (dMMR) EC that has progressed on or after a platinum containing regimen.
Patients received dostarlimab 500 mg every 3 weeks for 4 cycles followed by 1,000 mg every 6 weeks. Treatment continued until unacceptable toxicity or disease progression for up to two years. The major efficacy outcome measures were objective response rate (ORR) and duration of response (DOR) as assessed by blinded independent central radiologists' (BICR) review according to RECIST v1.1.
All patients included in both the primary and secondary efficacy analysis set had a minimum follow up period of 24 weeks from first dose, regardless of whether they had a post-treatment scan.
At the time of the third interim analysis (data cut-off 01 November 2021), a total of 141 patients with dMMR EC were evaluated for efficacy in the GARNET study. Among these 141 patients, the baseline characteristics were: median age 65.0 years (53% age 65 or older); 76.6% White, 3.5% Asian, 2.8% Black; and ECOG PS 0 (38.3%) or 1 (61.7%). The median number of prior therapies for recurrent or advanced endometrial cancer was one: 63% of patients had one prior line, 37% had two or more prior lines.
The identification of dMMR/MSI-H tumour status was prospectively determined based on local testing.
Local diagnostic assays (IHC, PCR or NGS) available at the sites were used for the detection of the dMMR/MSI-H expression in tumour material. Most of the sites used IHC as it was the most common assay available.
Efficacy results are shown in Table 9.

Elderly patients.
Of the 515 patients treated with dostarlimab monotherapy (IA1 GARNET population at time of data cut-off 01 March 2020), 51% were under 65 years, 38% were 65-75 years, and 12% were 75 years or older. No overall differences in safety or effectiveness were observed between patients under 65 years and in patients 65 years and older.
Paediatric population.
The safety and efficacy of dostarlimab in children and adolescents below 18 years of age have not been established.
5.2 Pharmacokinetic Properties
The pharmacokinetics (PK) of dostarlimab was assessed as monotherapy and when administered in combination with chemotherapy.
Dostarlimab PK as monotherapy or in combination with chemotherapy were characterised using population PK analysis from 869 patients with various solid tumours, including 546 patients with EC. The PK of dostarlimab are approximately dose proportional. When dosed at the recommended therapeutic dose for monotherapy (500 mg administered intravenously every 3 weeks for 4 doses, followed by 1,000 mg every 6 weeks), or at the recommended therapeutic dose for combination with chemotherapy (500 mg administered intravenously every 3 weeks for 6 doses, followed by 1000 mg every 6 weeks), dostarlimab shows an approximate two-fold accumulation (Cmin), consistent with the terminal half-life. The exposure of dostarlimab as monotherapy and/or in combination with chemotherapy was similar.
Absorption.
Dostarlimab is administered via the intravenous route and therefore estimates of absorption are not applicable.
Distribution.
The geometric mean volume of distribution of dostarlimab at steady state is approximately 5.81 L (CV% of 14.2%).
Metabolism.
Dostarlimab is a therapeutic mAb IgG4 that is expected to be catabolised into small peptides, amino acids, and small carbohydrates by lysosome through fluid-phase or receptor-mediated endocytosis. The degradation products are eliminated by renal excretion or returned to the nutrient pool without biological effects.
Excretion.
The geometric mean clearance is 0.00681 L/h (CV% of 30.2%) at steady state. The geometric mean terminal half-life (t1/2) at steady state is 23.2 days (CV% of 20.8%).
Dostarlimab clearance was estimated to be 7.8% lower when dostarlimab was given in combination with chemotherapy. There was no meaningful impact on dostarlimab exposure.
Special populations.
A population PK analysis of the patient data indicates that there are no clinically important effects of age (range: 24 to 86 years), sex or race, ethnicity, or tumour type on the clearance of dostarlimab. This population PK model also indicates that alterations in renal function (normal to moderate) and hepatic function (normal to mild impairment) do not alter the disposition of dostarlimab.
5.3 Preclinical Safety Data
Genotoxicity.
No studies have been performed to assess the potential of dostarlimab for genotoxicity.
Carcinogenicity.
No studies have been performed to assess the potential of dostarlimab for carcinogenicity.6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium citrate dihydrate, citric acid monohydrate, arginine hydrochloride, sodium chloride, polysorbate 80, water for injection.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store in a refrigerator 2°C to 8°C.
Do not freeze.
Store in the original carton until time of preparation in order to protect from light. The prepared dose may be stored either:
At room temperature up to 25°C for no more than 6 hours from the time of dilution until the end of infusion.
Under refrigeration at 2°C to 8°C for no more than 24 hours from time of dilution until end of infusion. If refrigerated, allow the diluted solution to come to room temperature prior to administration.
After preparation of infusion.
To reduce microbiological hazard, use as soon as practicable after reconstitution/preparation. If not used immediately, in-use chemical and physical stability have been demonstrated for up to 24 hours at 2°C to 8°C and up to 6 hours at room temperature (up to 25°C) from time of vial puncture to the end of administration.
Due to the lack of preservative, the product must not be used beyond these storage times.
Product is for single use in one patient only, discard any residue.
6.5 Nature and Contents of Container
10 mL Type I borosilicate clear glass vial, with a grey chlorobutyl elastomer stopper laminated with fluoropolymer, sealed with an aluminium flip-off cap containing 500 mg dostarlimab.
Each carton contains one vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
2022215-59-2.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
