
What is in this leaflet
This leaflet answers some common questions about Jinarc.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Jinarc is used for
Jinarc is used to treat a disease called Autosomal Dominant Polycystic Kidney Disease (ADPKD). This disease causes growth of cysts in the kidneys which results in problems because of their size and the space they occupy.
Jinarc contains the active ingredient tolvaptan which belongs to a group of medicines called vasopressin antagonists.
This means that it prevents a hormone called vasopressin from binding to receptors in your kidneys. By blocking the effect of vasopressin, Jinarc slows the development of kidney cysts in patients with ADPKD, reduces symptoms of the disease and increases urine production.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
This medicine is available only with a doctor’s prescription.
Jinarc is not recommended for use in children and teenagers, or patients older than 55 years. The effects of Jinarc in people younger than 18 years had not been studied, and in patients older than 55 years has not been proven.
Before you take Jinarc
When you must not take it
Do not take this medicine if:
- you have an allergy to tolvaptan, the active ingredient, benzazepine derivatives or to any of the other ingredients listed at the end of this leaflet under Product Description
- you have been told that you have raised levels of liver enzymes in your blood
- you have high level of sodium in your blood ("hypernatraemia")
- you do not realise when you are thirsty
- your kidneys cannot produce urine
- you have low blood volume
- you are pregnant
- you are breastfeeding
Do not take this medicine if you are below the age of 18 years. Safety and effectiveness in children younger than 18 years have not been established.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If the medicine has expired or the packing is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- you cannot drink enough water or if you are fluid restricted
- you have difficulties in urination or have an enlarged prostate
- you suffer from liver disease
- you suffer from too high or too low blood sodium
- you suffer from high potassium levels in your blood
- you have diabetes
- you have high blood pressure and are taking medications to treat it
- you are dehydrated or suffer from excessive vomiting, diarrhoea or sweating
- you have gout
- lactose intolerance
Tell your doctor if you are pregnant or plan to become pregnant or are breastfeeding. Jinarc must not be taken if you are pregnant or while breastfeeding.
You must use a reliable method of contraception to avoid becoming pregnant while you are taking Jinarc. You should continue doing this for one month after stopping treatment.
If you have not told your doctor about any of the above, tell him/her before you start taking Jinarc.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
In particular let your doctor know if you are taking:
- treatments containing ketoconazole, fluconazole or itraconazole for fungal infections
- antibiotics such as clarithromycin, erythromycin, and ciprofloxacin
- medicines for the treatment of HIV such as saquinavir, ritonavir and atazanavir
- medicines for high blood pressure and chest pain such as diltiazem and verapamil
- medicines which increase the level of sodium in your blood or which contain large amounts of salt, like tablets that dissolve in water and indigestion remedies
- digoxin, a medicine for the treatment of irregular heart beat and heart failure
- cyclosporine, a medicine that reduces the immune response
- quinidine, a medicine used for malaria
- dabigatran, a medicine used to thin the blood
- rosuvastatin and pitavastatin, medicines used to lower cholesterol
- methotrexate, a medicine used to treat rheumatoid arthritis
- sulfasalazine, a medicine used to treat inflammatory bowel disease
- metformin, a medicine for diabetes
- medicines for the treatment of epilepsy such as phenytoin and carbamazepine
- rifampicin, an antibiotic
- St John's wort, a traditional herbal medicinal product for the relief of slightly low mood and mild anxiety
- fluid tablets and other medicines used for the treatment of high blood pressure
- desmopressin, a medicine used to control urine output or bedwetting
It may still be alright for you to take these medicines and Jinarc together. Your doctor will be able to decide what is suitable for you.
Jinarc with food and drink
Do not drink grapefruit juice when taking Jinarc.
How to take Jinarc
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
How much to take
Jinarc is to be taken in two different doses every day. The dose combinations are 45 mg + 15 mg or 60 mg + 30 mg or 90 mg + 30 mg. One tablet of the higher dose (45 mg, 60 mg or 90 mg) should be taken in the morning upon waking, at least 30 minutes before food and one tablet with the lower dose (15 mg or 30 mg) should be taken 8 hours later. The afternoon dose can be taken with or without food.
Your doctor will start with a dose combination of 45 mg in the morning and 15 mg eight hours later and may then increase it to a maximum of 90 mg in the morning and 30 mg after 8 hours.
To find the best dosage for you your doctor will regularly examine whether you tolerate a prescribed dose. You should always take the highest tolerable dose combination prescribed by your doctor.
If you take other medicines which can increase the effects of Jinarc you may receive lower Jinarc doses.
Your doctor may have prescribed a different dose.
Ask your doctor or pharmacist if you are unsure of the correct dose for you. They will tell you exactly how much to take.
Follow the instructions they give you. If you take the wrong dose, Jinarc may not work as well and your problem may not improve.
How to take it
Swallow the tablets without chewing, with a glass of water.
The morning dose should be taken at least 30 minutes before the morning meal. The second daily dose can be taken with or without food.
Do not chew, crush or split the tablets. To ensure you get the entire dose, the tablets should be swallowed whole without chewing or crushing.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
Your first dose should be taken in the morning and your second dose approximately 8 hours later.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
Do not stop unless your doctor advises you to.
Your doctor may reduce your dose slowly to avoid side effects.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
If you forget to take it
If you forget to take your medicine you should take the dose as soon as you remember on the same day.
If you do not take your tablets on one day, take your normal dose on the next day. DO NOT take a double dose to make up for forgotten individual doses.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone Australia 13 11 26 for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Jinarc. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking Jinarc
Things you must do
MAKE SURE YOU DRINK ENOUGH WATER
Jinarc causes water loss because it increases your urine production. You may experience urine loss of between 5-7 L per day.
This water loss may result in side effects such as dry mouth and thirst or even more severe side effects like kidney problems (see "Side Effects"). It is therefore important that you have access to water and that you are able to drink sufficient amounts when you feel thirsty.
Unless your doctor tells you otherwise, drink plenty of water during the day and 1 or 2 glasses before going to bed, even if you do not feel thirsty and you must also drink water after you urinate at night.
Exposure to prolonged heat and humidity, exercise and intercurrent illness can further increase your risk of dehydration. In these circumstances, you should drink more water or fluid to reduce your risk of dehydration.
Special care must be taken if you have a disease that reduces appropriate fluid intake or if you are at an increased risk of water loss e.g. in case of vomiting or diarrhoea. If this occurs stop your tolvaptan treatment and seek medical advice immediately.
Due to the increased urine production it is also important that you always have access to a toilet.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Jinarc.
Tell any other doctors, dentists and pharmacists who treat you that you are taking Jinarc.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking Jinarc. It may affect other medicines used during surgery.
If you become pregnant while taking Jinarc, tell your doctor immediately. Your doctor will advise you on whether you should stop treatment. Do not stop treatment without first discussing it with your doctor.
If you are about to have any blood tests, tell your doctor that you are taking Jinarc. It may interfere with the results of some tests.
During treatment with Jinarc, your doctor will arrange regular (e.g. monthly) blood tests to check for changes in your liver function.
Keep all of your doctor’s appointments so that your progress can be checked.
Things you must not do
Do not take Jinarc to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
Patients starting Jinarc should be carefully observed especially when starting treatment and if the dose is increased.
Do not drive or operating machinery until you know how Jinarc affects you. Jinarc may cause side effects that can affect your ability to drive or use machines.
Jinarc may make you feel dizzy or sleepy, particularly at the beginning of treatment. If this happens to you, do not drive or use any tools or machines.
Side effects
Serious side effects:
If you notice any of the following side effects, you may need urgent medical attention. Stop taking Jinarc and immediately contact a doctor or go to the nearest hospital if you:
- find it difficult to urinate
- experience swelling of the face, lips or tongue, itching, generalised rash, or severe wheezing or breathlessness (symptoms of an allergic reaction)
- signs of electrolytes imbalances such as dizziness, confusion, weakness, seizures or palpitation
Jinarc may cause your liver to not work properly. Therefore, please inform your doctor immediately if you have any signs that could indicate potential liver problems, such as:
- nausea
- vomiting
- fever
- tiredness
- loss of appetite
- pain in the abdomen
- dark urine
- jaundice (yellowing of the skin or eyes)
- itching of your skin
- joint and muscle pain with fever
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Jinarc.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them:
- thirst
- increased amount of urine
- increased frequency of urination during the day and at night
- headache
- constipation, diarrhea, dry mouth, indigestion, decreased appetite
- fatigue, weakness, dizziness
- trouble sleeping
- muscle spasms
- rash, dry skin, itching
- painful, swollen joints
If any of these affects you severely, tell your doctor, nurse or pharmacist.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell, even if it is not listed in this leaflet. Other side effects not listed above may also occur in some people.
Ask your doctor or pharmacist to answer any questions you may have.
After using Jinarc
Storage
Keep your medicine in the original container.
If you take it out of its original container it may not keep well.
Keep your medicine in a cool dry place where the temperature stays below 25°C.
Do not store Jinarc or any other medicine in the bathroom or near a sink.
Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
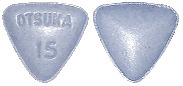
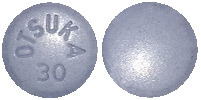
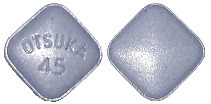
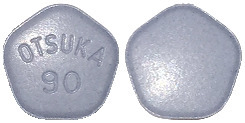
The different strengths of Jinarc tablets have different shapes and embossing:
15 mg tablet: blue, triangular, debossed with "OTSUKA" and "15" on one side.
30 mg tablet: blue, round, debossed with "OTSUKA" and "30" on one side.
45 mg tablet: blue, square, debossed with "OTSUKA" and "45" on one side
60 mg tablet: blue, modified rectangular, debossed with "OTSUKA" and "60" on one side
90 mg tablet: blue, pentagonal, debossed with "OTSUKA" and "90" on one side
Ingredients
Active ingredient:
tolvaptan
Excipient Ingredients:
- lactose monohydrate
- maize starch
- microcrystalline cellulose
- hyprolose
- magnesium stearate
- indigo carmine aluminium lake.
This medicine does not contain gluten, tartrazine or any other azo dyes.
Sponsor
Otsuka Australia Pharmaceutical Pty Ltd
Suite 2.03, Level 2
9 Help Street, Chatswood NSW 2067
Under the licence of Otsuka Pharmaceutical Co., Ltd.
Date of Preparation
June 2020
Australian Register Numbers
AUST R 272785 - 15 mg tablets blister pack
AUST R 272786 - 30 mg tablet blister pack
AUST R 272787 - 15 mg + 45 mg tablet blister composite pack
AUST R 272788 - 30 mg + 60 mg tablet blister composite pack
AUST R 272789 = 30 mg + 90 mg tablet blister composite pack
Published by MIMS August 2020

 Further reductions have to be considered if patients cannot tolerate the reduced tolvaptan doses.
Further reductions have to be considered if patients cannot tolerate the reduced tolvaptan doses.

 The next sequentially ordered secondary endpoint of slope of kidney function decline was assessed as change in estimated glomerular filtration rate (eGFR-CKD EPI) during treatment (from end of titration to last on-drug visit). The tolvaptan-treated patients had a 26.4% reduction in the rate of renal function decline compared with placebo (-2.7 versus -3.7 (mL/min/1.73 m2), p < 0.0001, Figure 2). Figure 2 represents slope of renal function for tolvaptan (solid) and placebo (dashed) change from end of titration baseline (i.e. end of Week 3). Box plots derived from mixed effect model repeat measurement (MMRM) analyses to each indicated 12-month visit with 5th, 25th, mean, 75th and 95th percentiles of change from end of titration for tolvaptan (grey) and placebo (white) groups.
The next sequentially ordered secondary endpoint of slope of kidney function decline was assessed as change in estimated glomerular filtration rate (eGFR-CKD EPI) during treatment (from end of titration to last on-drug visit). The tolvaptan-treated patients had a 26.4% reduction in the rate of renal function decline compared with placebo (-2.7 versus -3.7 (mL/min/1.73 m2), p < 0.0001, Figure 2). Figure 2 represents slope of renal function for tolvaptan (solid) and placebo (dashed) change from end of titration baseline (i.e. end of Week 3). Box plots derived from mixed effect model repeat measurement (MMRM) analyses to each indicated 12-month visit with 5th, 25th, mean, 75th and 95th percentiles of change from end of titration for tolvaptan (grey) and placebo (white) groups. Subgroup analysis of all endpoints above (change in TKV, key composite [including time to worsening of kidney function and time to medically significant kidney pain] and change in slope of decline in renal function) demonstrated consistent efficacy (directional) in all pre-specified subgroups, including those stratified by age, gender, race, geographical location, baseline hypertension, baseline eGFR and baseline TKV.
Subgroup analysis of all endpoints above (change in TKV, key composite [including time to worsening of kidney function and time to medically significant kidney pain] and change in slope of decline in renal function) demonstrated consistent efficacy (directional) in all pre-specified subgroups, including those stratified by age, gender, race, geographical location, baseline hypertension, baseline eGFR and baseline TKV. The key secondary endpoint was a comparison of the efficacy of tolvaptan treatment versus placebo in reducing the decline of annualised eGFR slope across all measured time points in the trial. These data also showed statistically significant benefit from tolvaptan vs. placebo (p < 0.0001) (Table 6).
The key secondary endpoint was a comparison of the efficacy of tolvaptan treatment versus placebo in reducing the decline of annualised eGFR slope across all measured time points in the trial. These data also showed statistically significant benefit from tolvaptan vs. placebo (p < 0.0001) (Table 6). Prespecified subgroup analyses of the primary endpoint showed a beneficial effect of tolvaptan across subgroups that were defined according to sex, baseline estimated GFR, stage of chronic kidney disease (except for stage 2) and geographic region, as well as in the subgroups of patients who were 55 years of age or younger and patients who were white.
Prespecified subgroup analyses of the primary endpoint showed a beneficial effect of tolvaptan across subgroups that were defined according to sex, baseline estimated GFR, stage of chronic kidney disease (except for stage 2) and geographic region, as well as in the subgroups of patients who were 55 years of age or younger and patients who were white. Data are not currently available to show whether long-term therapy with Jinarc continues to slow the rate of renal function decline and affect clinical outcomes of ADPKD, including delay in the onset of end-stage renal disease.
Data are not currently available to show whether long-term therapy with Jinarc continues to slow the rate of renal function decline and affect clinical outcomes of ADPKD, including delay in the onset of end-stage renal disease. Chemical Name: (±)-4'-[(7-chloro- 2,3,4,5-tetrahydro -5-hydroxy-1H-1-benzazepin -1-yl) carbonyl]-o-tolu-m-toluidide.
Chemical Name: (±)-4'-[(7-chloro- 2,3,4,5-tetrahydro -5-hydroxy-1H-1-benzazepin -1-yl) carbonyl]-o-tolu-m-toluidide.