1 Name of Medicine
Finerenone.
2 Qualitative and Quantitative Composition
Kerendia tablets contain 10 mg, 20 mg or 40 mg of finerenone.
Excipients with known effect.
Contains lactose.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Kerendia 10 mg film-coated tablet.
Pink, oval-oblong tablet with a length of 10 mm and a width of 5 mm, marked '10' on one side and 'FI' on the other side.
Kerendia 20 mg film-coated tablet.
Pale-yellow, oval-oblong tablet with a length of 10 mm and a width of 5 mm, marked '20' on one side and 'FI' on the other side.
Kerendia 40 mg film-coated tablet.
Grey-orange, oval-oblong tablet with a length of 11 mm and a width of 5 mm, marked '40' on one side and 'FI' on the other side.4.1 Therapeutic Indications
Chronic kidney disease.
Kerendia is indicated to delay progressive decline of kidney function and to reduce the risk of cardiovascular mortality and morbidity in adults with chronic kidney disease (with albuminuria) associated with type 2 diabetes, in addition to standard of care (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Heart failure.
Kerendia is indicated in adults for the treatment of symptomatic heart failure with left ventricular ejection fraction (LVEF) of ≥ 40%.4.2 Dose and Method of Administration
Dosage (dose and interval).
Initiation of treatment. Serum potassium limits. Chronic kidney disease.
Initiation of Kerendia treatment is recommended when serum potassium ≤ 4.8 mmol/L. If serum potassium > 4.8 to 5.0 mmol/L, initiation of Kerendia treatment may be considered with additional serum potassium monitoring within the first 4 weeks based on patient characteristics and serum potassium levels. If serum potassium > 5.0 mmol/L, initiation of Kerendia treatment is not recommended (see Section 4.4 Special Warnings and Precautions for Use).
Heart failure.
Initiation of Kerendia treatment is recommended when serum potassium ≤ 5.0 mmol/L (see Section 4.4 Special Warnings and Precautions for Use).
For monitoring of serum potassium, see Continuation of treatment.
Starting dose for all indications. Measure estimated glomerular filtration rate (eGFR) to determine the starting dose. The starting dose of Kerendia is:
20 mg once daily if eGFR ≥ 60 mL/min/1.73 m2;
10 mg once daily if eGFR ≥ 25 to < 60 mL/min/1.73 m2.
Initiation of Kerendia treatment is not recommended in patients with eGFR < 25 mL/min/1.73 m2 as clinical experience is limited.
Target dose. Chronic kidney disease.
The recommended target dose of Kerendia is 20 mg once daily.
Heart failure.
The recommended target dose of Kerendia depends on renal function (eGFR) at initiation of Kerendia treatment:
40 mg once daily if eGFR ≥ 60 mL/min/1.73 m2;
20 mg once daily if eGFR ≥ 25 to < 60 mL/min/1.73 m2.
Continuation of treatment. Four weeks after initiation or re-start or dose adjustment of Kerendia treatment, re-measure serum potassium and eGFR. See Tables 1 or 2 to determine continuation of Kerendia treatment and dose adjustment. Thereafter, remeasure serum potassium periodically and as needed based on patient characteristics and serum potassium levels. (See Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).


Method of administration.
Oral use.
Tablets may be taken with a glass of water and with or without food (see Section 5.2 Pharmacokinetic Properties).
Avoid taking Kerendia with grapefruit or grapefruit juice (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
For patients who are unable to swallow whole tablets, Kerendia tablet may be crushed and mixed with water or soft foods, such as apple sauce, immediately prior to use and administered orally (see Section 5.2 Pharmacokinetic Properties).
Missed dose.
A missed dose should be taken as soon as possible after it is noticed, but only on the same day. If this is not possible, the dose should be skipped and the next dose taken as prescribed. Two doses should not be taken to make up for a missed dose.
Maximum daily dose.
Chronic kidney disease.
The maximum daily dose of Kerendia is 20 mg.
Heart failure.
The maximum daily dose of Kerendia is 40 mg.
Patients with hepatic impairment.
In patients with severe hepatic impairment (Child-Pugh C), avoid treatment with Kerendia (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties). In patients with mild or moderate hepatic impairment, no initial dose adjustment is required (Child-Pugh A or B) (see Section 5.2 Pharmacokinetic Properties).
In patients with moderate hepatic impairment (Child-Pugh B), consider additional serum potassium monitoring and adapt monitoring according to patient characteristics (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Patients with renal impairment.
Initiation of Kerendia treatment.
In patients with eGFR ≥ 25 to < 60 mL/min/1.73 m2, the starting dose of Kerendia is 10 mg once daily. See Initiation of treatment.
In patients with eGFR < 25 mL/min/1.73 m2, initiation of Kerendia treatment is not recommended as clinical experience is limited (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Continuation of Kerendia treatment.
In patients with mild, moderate or severe renal impairment, continue Kerendia treatment and adjust dose based on serum potassium. Measure eGFR 4 weeks after initiation to determine the dose adjustment. See Tables 1 or 2, Continuation of treatment.
In patients with end-stage renal disease (eGFR < 15 mL/min/1.73 m2), continue Kerendia treatment with caution regarding serum potassium levels as clinical experience is limited (see Section 4.4 Special Warnings and Precautions for Use).
Patients taking concomitant medications.
In patients taking Kerendia concomitantly with moderate or weak CYP3A4 inhibitors, potassium supplements, trimethoprim, or trimethoprim-sulfamethoxazole, consider additional serum potassium monitoring and adapt monitoring according to patient characteristics, and make Kerendia treatment decisions as directed in Tables 1 or 2. Temporary discontinuation of Kerendia when taking trimethoprim, or trimethoprim-sulfamethoxazole, may be necessary (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Elderly patients.
No dose adjustment is required in the elderly (see Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
Kerendia is contraindicated in patients:
taking concomitant medications that are strong CYP3A4 inhibitors (e.g. itraconazole, ketoconazole, ritonavir, nelfinavir, cobicistat, clarithromycin, telithromycin and nefazodone) (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions);
with adrenal insufficiency.
4.4 Special Warnings and Precautions for Use
Hyperkalaemia.
Hyperkalaemia has been observed in patients treated with Kerendia. Some patients are at a higher risk to develop hyperkalaemia. Risk factors include low eGFR, higher serum potassium and previous episodes of hyperkalaemia. Consider more frequent monitoring in these patients.
Initiation of Kerendia treatment is not recommended if serum potassium > 5.0 mmol/L.
Chronic kidney disease.
If serum potassium > 4.8 to 5.0 mmol/L, initiation of Kerendia treatment may be considered with additional serum potassium monitoring within the first 4 weeks based on patient characteristics and serum potassium levels (see Section 4.2 Dose and Method of Administration).
In treated patients with chronic kidney disease, withhold Kerendia if serum potassium > 5.5 mmol/L. Follow local guidelines for the management of hyperkalaemia. Restart Kerendia at 10 mg once daily if serum potassium ≤ 5.0 mmol/L (see Section 4.2 Dose and Method of Administration).
Heart failure.
Withhold Kerendia if serum potassium ≥ 6.0 mmol/L. Follow local guidelines for the management of hyperkalaemia. Restart Kerendia at 10 mg once daily if serum potassium < 5.5 mmol/L or if repeatedly ≥ 5.5 mmol/L, wait to restart until < 5.0 mmol/L (see Section 4.2 Dose and Method of Administration).
In all patients, re-measure serum potassium and eGFR 4 weeks after initiation or re-start or dose adjustment of Kerendia treatment. Thereafter, remeasure serum potassium periodically and as needed based on patient characteristics and serum potassium levels (see Section 4.2 Dose and Method of Administration).
Worsening of renal function in patients with heart failure.
Kerendia may cause an initial decrease in eGFR due to hemodynamic changes related to the mechanism of action of the drug. This decrease is typically observed within weeks of initiating treatment and then stabilizes and is reversible after treatment discontinuation.
In patients with heart failure, a greater risk of events related to worsening of renal function was observed in patients receiving Kerendia treatment compared to placebo. Most of these events were mild to moderate in severity. Rarely, severe events associated with worsening renal function, including events requiring hospitalization, have been observed [see Section 4.8 Adverse Effects (Undesirable Effects), Heart failure (LVEF ≥ 40%)].
Measure eGFR to guide treatment initiation and dosage adjustment (see Section 4.2 Dose and Method of Administration). Consider delaying up-titration or interrupting treatment with Kerendia in patients who develop clinically significant worsening of renal function.
Concomitant medications.
The risk of hyperkalaemia also may increase with the intake of concomitant medications that may increase serum potassium (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Also see Concomitant use of substances that affect finerenone exposure.
Avoid concomitant use of Kerendia with the following medications:
potassium-sparing diuretics (e.g. amiloride, triamterene);
other mineralocorticoid receptor antagonists (MRAs) (e.g. eplerenone, spironolactone).
Use Kerendia with caution and monitor serum potassium when taken concomitantly with the following medications:
potassium supplements;
trimethoprim, or trimethoprim-sulfamethoxazole. Temporary discontinuation of Kerendia may be necessary.
Use in hepatic impairment.
Patients with severe hepatic impairment (Child-Pugh C) have not been studied (see Section 5.2 Pharmacokinetic Properties). Due to an expected significant increase in finerenone exposure, avoid use of Kerendia in patients with severe hepatic impairment (see Section 4.2 Dose and Method of Administration).
Due to an increase in finerenone exposure, consider additional serum potassium monitoring and adapt monitoring according to patient characteristics in patients with moderate hepatic impairment (Child-Pugh B) (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Use in renal impairment.
The risk of hyperkalaemia increases with decreasing renal function. Ongoing monitoring of renal function should be performed as needed according to standard practice (see Section 4.2 Dose and Method of Administration).
Initiation of Kerendia treatment is not recommended in patients with eGFR < 25 mL/min/1.73 m2 as clinical experience is limited (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Continue Kerendia treatment with caution regarding serum potassium levels in patients with end-stage renal disease (eGFR < 15 mL/min/1.73 m2) as clinical experience is limited (see Section 4.2 Dose and Method of Administration).
Use in the elderly.
No dose adjustment is required in the elderly (see Section 5.2 Pharmacokinetic Properties).
Paediatric use.
The safety and efficacy of Kerendia have not been established in patients under 18 years of age. Therefore, Kerendia is not recommended for use in paediatric patients.
Effects on laboratory tests.
See Section 4.8 Adverse Effects (Undesirable Effects); Table 5: Laboratory test abnormalities reported in ≥ 1% of patients on Kerendia and more frequently than placebo in phase III studies (pooled FIDELIO-DKD, FIGARO-DKD, and FINEARTS-HF); Table 3: Adverse reactions reported with Kerendia in phase III studies (pooled FIDELIO-DKD, FIGARO-DKD,
and FINEARTS-HF); Section 5.1 Pharmacodynamic Properties, Clinical trials.
Embryo-fetal toxicity.
Animal data have shown reproductive toxicity. The relevance for humans is unknown. Kerendia should not be used during pregnancy unless there has been careful consideration of the benefit for the mother and the risk to the fetus. If the patient becomes pregnant while taking Kerendia, the patient should be informed of potential risks to the fetus. Advise women of childbearing potential to use effective contraception during treatment with Kerendia. Advise women not to breastfeed during treatment with Kerendia (see Section 4.6 Fertility, Pregnancy and Lactation).
Concomitant use of substances that affect finerenone exposure.
Moderate and weak CYP3A4 inhibitors.
The concomitant use of Kerendia with moderate CYP3A4 inhibitors (e.g. erythromycin and verapamil) and weak CYP3A4 inhibitors (e.g. amiodarone and fluvoxamine) is expected to increase finerenone exposure (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Monitor serum potassium especially during initiation of or changes to dosing of Kerendia or the CYP3A4 inhibitor (see Section 4.2 Dose and Method of Administration).
Strong and moderate CYP3A4 inducers.
Avoid concomitant use of Kerendia with strong CYP3A4 inducers (e.g. rifampicin, carbamazepine, phenytoin, phenobarbital, St John's wort) or moderate CYP3A4 inducers (e.g. efavirenz), which are expected to markedly decrease finerenone plasma concentrations and result in reduced therapeutic effect (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Consider selection of an alternate concomitant medicinal product with no or weak potential to induce CYP3A4.
Grapefruit.
Avoid concomitant intake of grapefruit or grapefruit juice as it is expected to increase the plasma concentration of finerenone (see Section 4.2 Dose and Method of Administration; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).4.5 Interactions with Other Medicines and Other Forms of Interactions
Effects of other substances on finerenone.
Finerenone is cleared almost exclusively via cytochrome P450 (CYP)-mediated oxidative metabolism (mainly CYP3A4 [90%] with a small contribution of CYP2C8 [10%]). Accordingly, agents that affect CYP3A4 may significantly alter exposure to finerenone.
Effect of CYP3A4 inhibitors on finerenone.
Strong CYP3A4 inhibitors.
Simulations suggest that concomitant use of Kerendia with itraconazole (200 mg BID), a strong CYP3A4 inhibitor, increases finerenone AUC (+531%) and Cmax (+137%). Clarithromycin (500 mg BID), another strong inhibitor, also is predicted to increase finerenone AUC (+428%) and Cmax (+125%). Due to an expected marked increase in finerenone exposure, concomitant use of Kerendia with itraconazole, clarithromycin and other strong CYP3A4 inhibitors (e.g. ketoconazole, ritonavir, nelfinavir, cobicistat, telithromycin or nefazodone) is contraindicated (see Section 4.3 Contraindications).
Moderate CYP3A4 inhibitors.
Concomitant use of erythromycin (500 mg thrice daily), a moderate CYP3A4 inhibitor, increased finerenone mean AUC and Cmax by 248% and 88%, respectively. Another moderate CYP3A4 inhibitor, verapamil (240 mg controlled-release tablet once daily), increased finerenone mean AUC and Cmax by 170% and 120%, respectively. Serum potassium may increase, and therefore, monitoring of serum potassium is recommended (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Weak CYP3A4 inhibitors.
In an analysis of Kerendia in patients, the use of amiodarone, a weak CYP3A4 inhibitor, was estimated to result in a 21% increase of finerenone AUC. Simulations suggest that fluvoxamine (100 mg BID), another weak inhibitor, increases finerenone AUC (+57%) and Cmax (+38%). Serum potassium may increase, and therefore, monitoring of serum potassium is recommended (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Grapefruit.
Concomitant intake of grapefruit or grapefruit juice is expected to increase the plasma concentration of finerenone and should be avoided (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Effect of strong and moderate CYP3A4 inducers on finerenone.
Simulations suggest that rifampicin (600 mg OD), a strong CYP3A4 inducer, decreases finerenone AUC (-93%) and Cmax (-86%). Efavirenz (600 mg OD), a moderate CYP3A4 inducer, is predicted to decrease finerenone AUC (-81%) and Cmax (-68%).
Concomitant use of Kerendia with rifampicin and other strong CYP3A4 inducers (e.g. carbamazepine, phenytoin, phenobarbital, St John's wort) or with efavirenz and other moderate CYP3A4 inducers, markedly decreases finerenone plasma concentration and results in reduced therapeutic effect and should be avoided (see Section 4.4 Special Warnings and Precautions for Use).
Lack of clinically relevant drug-drug interaction.
Concomitant use of gemfibrozil (600 mg twice daily), a strong inhibitor of CYP2C8, increased finerenone mean AUC and Cmax by 10% and 16%, respectively. This is not clinically relevant.
Pre- and co-treatment with the proton pump inhibitor omeprazole (40 mg once daily) had no effect on finerenone mean AUC and mean Cmax.
Concomitant use of antacid aluminium hydroxide and magnesium hydroxide (70 mVal) had no effect on finerenone mean AUC and reduced its mean Cmax by 19%. This is not clinically relevant.
Effect of finerenone on other substances.
At 40 mg once daily, finerenone is a weak inhibitor of the CYP3A4 enzyme in vivo. Coadministration of multiple doses of 40 mg finerenone with the CYP3A4 probe substrate midazolam resulted in a mean AUC increase of 31% with no effect on Cmax. Consider the potential increased exposure of sensitive CYP3A4 substrates with a narrow therapeutic window in case of concomitant use of finerenone 40 mg once daily. A multiple-dose regimen of 20 mg finerenone once-daily had no effect on the AUC of the CYP3A4 probe substrate midazolam. Finerenone neither inhibits nor induces CYP3A4 at this dose level.
At 40 mg once daily, finerenone is a weak inhibitor of the CYP2C8 enzyme in vivo. Coadministration of multiple doses of 40 mg finerenone with the CYP2C8 probe substrate repaglinide resulted in a mean AUC and Cmax increase of 59% and 30%, respectively. Consider the potential increased exposure of sensitive CYP2C8 substrates with a narrow therapeutic window in case of concomitant use of finerenone 40 mg once daily. A single dose of 20 mg finerenone had no effect on AUC and Cmax of the CYP2C8 probe substrate repaglinide. Finerenone does not inhibit CYP2C8 at this dose level.
Lack of mutual pharmacokinetic interaction was demonstrated between finerenone and the CYP2C9 substrate warfarin and between finerenone and the P-gp substrate digoxin.
Multiple doses of 40 mg finerenone once daily had no clinically relevant effect on AUC or Cmax of the BCRP and OATP substrate rosuvastatin.
Pharmacodynamic interactions.
Medications that increase serum potassium.
It is anticipated that medications that increase serum potassium will increase the risk of hyperkalaemia when used concomitantly with Kerendia.
Concomitant use of Kerendia with the following medications should be avoided:
potassium-sparing diuretics (e.g. amiloride, triamterene);
other mineralocorticoid receptor antagonists (MRAs) (e.g. eplerenone, spironolactone).
Kerendia should be used with caution and serum potassium monitored when taken concomitantly with the following medications:
potassium supplements;
trimethoprim, or trimethoprim-sulfamethoxazole. Temporary discontinuation of Kerendia may be necessary.
(See Section 4.4 Special Warnings and Precautions for Use.)4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No human data on the effect of Kerendia on fertility are available. No effect on male fertility was observed with finerenone in rats at oral doses up to 30 mg/kg/day (estimated to yield 16 or 6.2 times the exposure in patients at the maximum recommended human dose of 20 or 40 mg/day, respectively [based on plasma AUC for unbound drug]). In female rats, inhibition of ovulation was seen with oral treatment at 30 mg/kg/day (yielding 21 or 8.5 times the clinical exposure following a maximum recommended human dose of 20 or 40 mg/day, respectively), while no effect on female fertility was observed at 3 mg/kg/day (relative exposure, 10 or 4 following a maximum recommended human dose of 20 or 40 mg/day, respectively). Based on these animal data and the margin of exposure, impairment of male and female fertility is not expected in patients.
(Category D)
There are no data on the use of Kerendia in pregnant women.
Adverse effects on embryofetal development, including teratogenicity, were observed with finerenone in animals. Placental transfer of finerenone and/or its metabolites was demonstrated in the rat.
Administration of finerenone to pregnant rats reduced fetal weight and impaired fetal ossification at oral doses ≥ 10 mg/kg/day. Increased external and skeletal variations (slight oedema, shortened umbilical cord and slightly enlarged fontanelle) and malformation of the aortic arch were observed at 30 mg/kg/day. These doses yield exposure 19-25 or 7-10 times higher than in patients at the maximum recommended human dose of 20 or 40 mg/day, respectively (based on plasma AUC for unbound drug) and were maternotoxic. Maternal dosing with finerenone at oral doses ≥ 3 mg/kg/day during gestation and lactation (relative exposure, 4 or 1.7 at the maximum recommended human dose of 20 or 40 mg/day, respectively), decreased birth weight, increased perinatal mortality and slightly increased locomotor activity of rat pups. At 10 mg/kg/day, postnatal body weight gain was reduced and development delayed. The observed effect on neurobehaviour is consistent with a pharmacologically-mediated antidepressant-like effect of finerenone as a result of exposure in the fetal brain in utero. No adverse effects on embryofetal development were observed with finerenone in rabbits up to the highest dose tested (2.5 mg/kg/day, yielding 13 or 15 times the clinical exposure at the maximum recommended human dose of 20 or 40 mg/day, respectively).
Kerendia should not be used during pregnancy unless there has been careful consideration of the benefit for the mother and the risk to the fetus (see Section 4.4 Special Warnings and Precautions for Use).
Women of childbearing potential/ contraception.
Kerendia may cause embryofetal harm when administered during pregnancy. Women of childbearing potential should use effective contraception during treatment with Kerendia (see Section 4.4 Special Warnings and Precautions for Use).
It is unknown whether finerenone or its metabolites are excreted in human breast milk. Excretion of finerenone and its metabolites in milk was shown in rats. Rat pups exposed to finerenone in utero and through consumption of maternal milk showed adverse effects (see Section 4.6 Fertility, Pregnancy and Lactation). A risk to the nursing infant cannot be excluded. Breastfeeding should be discontinued if use of Kerendia is considered essential (see Section 4.4 Special Warnings and Precautions for Use).4.7 Effects on Ability to Drive and Use Machines
No effects on ability to drive and use machines have been observed.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
The safety of Kerendia in patients with chronic kidney disease and type 2 diabetes was evaluated in two pivotal phase III studies FIDELIO-DKD and FIGARO-DKD. In the FIDELIO-DKD study, 2,818 patients received Kerendia (10 or 20 mg once daily) with a mean duration of treatment of 2.2 years. In the FIGARO-DKD study, 3671 patients received Kerendia (10 mg or 20 mg once daily) with a mean duration of treatment of 2.9 years.
The safety of Kerendia in patients with heart failure (LVEF ≥ 40%) was evaluated in the phase III study, FINEARTS-HF. In this study, 2,993 patients received Kerendia (10 mg, 20 mg, or 40 mg once daily) with a mean duration of treatment of 2.1 years.
The most frequently reported (≥ 10%) adverse reaction was hyperkalaemia. See Description of selected adverse reactions below (see Section 4.4 Special Warnings and Precautions for Use).
Tabulated list of adverse reactions.
The adverse reactions reported with Kerendia are summarised in Table 3 by MedDRA system organ class and by frequency.
Adverse reactions are ranked by system organ class and then by frequency with the most frequent first, using the following convention: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).
Within each frequency grouping, adverse reactions are ranked in order of decreasing seriousness.
 The data in Table 4 and Table 5 reflect frequencies of Kerendia-ADRs from the pooled Phase III studies of FIGARO-DKD, FIDELIO-DKD and FINEARTS-HF with safety population of a total of 9482 subjects who were exposed to Kerendia and 9467 subjects on placebo.
The data in Table 4 and Table 5 reflect frequencies of Kerendia-ADRs from the pooled Phase III studies of FIGARO-DKD, FIDELIO-DKD and FINEARTS-HF with safety population of a total of 9482 subjects who were exposed to Kerendia and 9467 subjects on placebo.


Heart failure (LVEF ≥ 40%).
In the FINEARTS-HF study, the overall safety profile of Kerendia was largely consistent with the adverse events reported in patients with CKD and T2D; however, adverse events related to worsening of renal function were reported more frequently in the Kerendia group compared with placebo. The most frequently reported adverse events related to worsening of renal function included renal impairment (6.6% vs. 3.9%), eGFR decreased (5.2% vs. 3.6%), acute kidney injury (3.7% vs. 2.1%), renal failure (2.6% vs. 1.6%) and blood creatinine increased (1.2% vs. 0.8%). The majority of these events were mild to moderate. Events leading to permanent discontinuation were the same in patients receiving Kerendia or placebo (0.3%). Hospitalization due to events related to worsening of renal function in the Kerendia group was 2.0% versus 1.3% in the placebo group.
Description of selected adverse reactions.
Hyperkalaemia.
In the FIDELIO-DKD study including patients with CKD (mean eGFR 44.4 mL/min/1.73 m2) and type 2 diabetes, hyperkalaemia events were reported in 18.2% of Kerendia-treated patients compared with 9.0% of placebo-treated patients. An increase from baseline in mean serum potassium in the first month of treatment of approximately 0.2 mmol/L was observed in the Kerendia group compared to placebo, which remained stable thereafter. In the FIGARO-DKD study including patients with CKD (mean eGFR 67.8 mL/min/1.73 m2) and type 2 diabetes, hyperkalaemia events were reported in 10.7% of Kerendia-treated patients compared with 5.3% of placebo-treated patients. An increase from baseline in mean serum potassium in the first month of treatment of approximately 0.15 mmol/L was observed in the Kerendia group compared to placebo, which remained stable thereafter.
In the FINEARTS-HF study, including patients with heart failure (LVEF ≥ 40%), hyperkalaemia events were reported in 9.7% of Kerendia-treated patients compared with 4.2% of placebo-treated patients. An increase from baseline in mean serum potassium in the first month of treatment of approximately 0.2 mmol/L was observed in the Kerendia group compared to placebo, which remained stable thereafter.
In all studies, the majority of hyperkalaemia events were mild to moderate in patients treated with Kerendia. For specific recommendations, see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
No cases of adverse events associated with finerenone overdose in humans have been reported. The most likely manifestation of overdose is anticipated to be hyperkalaemia. If hyperkalaemia develops, standard treatment should be initiated.
Finerenone is unlikely to be efficiently removed by haemodialysis given its fraction bound to plasma proteins of about 90%.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
ATC code: C03DA05.
Mechanism of action.
Finerenone is a nonsteroidal antagonist of the mineralocorticoid receptor (MR) that potently attenuates inflammation and fibrosis mediated by MR overactivation. The MR is expressed in the kidneys, heart and blood vessels where finerenone also counteracts sodium retention and hypertrophic processes. Finerenone has high selectivity for the MR due to its nonsteroidal structure and bulky binding mode. Finerenone has no relevant affinity for androgen, progesterone, estrogen and glucocorticoid receptors and therefore does not cause sex hormone-related adverse events (e.g. gynecomastia). Its binding to the MR leads to a specific receptor ligand complex that blocks recruitment of transcriptional coactivators implicated in the expression of pro-inflammatory and pro-fibrotic mediators.
Effects in healthy participants.
Multiple dose regimens of finerenone (daily doses of 20 mg or 40 mg over 10 days) led to activation of the renin-angiotensin-aldosterone system (RAAS), i.e. reversible increases of plasma renin activity and serum aldosterone concentrations with baseline values reached again within 48 hours after the last dose.
Following activation of the MR with the agonist fludrocortisone single doses of finerenone up to 20 mg showed dose dependent natriuretic effects while decreasing urinary potassium excretion as compared to placebo.
Single or multiple doses of finerenone did not influence vital signs parameters in healthy participants.
Effects on UACR.
In FIDELIO-DKD and FIGARO-DKD, randomised, double-blind, placebo-controlled, multicentre phase III studies in adults with CKD and type 2 diabetes, the placebo-corrected relative reduction in urinary albumin-to-creatinine ratio (UACR) in patients randomised to finerenone at Month 4 was 31% and 32%, respectively and UACR remained reduced throughout both studies.
In FINEARTS-HF, a randomised, double-blind, placebo-controlled, multicentre phase III study in adults with heart failure (LVEF ≥ 40%), the placebo-corrected relative reduction in UACR in patients randomised to finerenone at month 6 was 30%, and UACR remained reduced up to the last measurement at year 2.
In ARTS-DN, a randomised, double-blind, placebo-controlled, multicentre phase IIb dose-finding study in adults with CKD and type 2 diabetes, the placebo-corrected relative reduction in UACR at Day 90 was 25% and 38% in patients treated with finerenone 10 mg and 20 mg once daily, respectively.
Cardiac electrophysiology.
In a thorough QT study in 57 healthy participants, there was no indication of a QT/QTc prolonging effect of finerenone after single doses of 20 mg (therapeutic) or 80 mg (supratherapeutic), indicating that finerenone has no effect on cardiac repolarization.
Clinical trials.
Chronic kidney disease. Kerendia was investigated in two randomised, double-blind, placebo-controlled, multicentre phase III studies, FIDELIO-DKD and FIGARO-DKD. In these studies, the effect of Kerendia on kidney and cardiovascular outcomes was evaluated in adults with CKD and type 2 diabetes receiving either Kerendia 10 mg or 20 mg once daily, or placebo.
In FIDELIO-DKD patients were eligible based on evidence of persistent albuminuria (> 30 mg/g to 5,000 mg/g), an eGFR of 25 to 75 mL/min/1.73 m2, serum potassium ≤ 4.8 mmol/L at screening, and were required to be receiving standard of care, including a maximum tolerated labelled dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB).
The primary endpoint in the FIDELIO-DKD study was a composite of time to first occurrence of kidney failure (defined as chronic dialysis or kidney transplantation, or a sustained decrease in eGFR to < 15 mL/min/1.73 m2 over at least 4 weeks), a sustained decline in eGFR of 40% or more compared to baseline over at least 4 weeks, or renal death. The key secondary endpoint was a composite of time to first occurrence of cardiovascular (CV) death, non-fatal myocardial infarction (MI), non-fatal stroke or hospitalisation for heart failure.
The trial analysed 5,662 patients randomly assigned to receive either Kerendia (N = 2824), or placebo (N = 2838), with a median follow-up duration of 2.6 years. After the end of study notification, vital status was obtained for 99.7% of patients. The trial population was 63% White, 25% Asian and 5% Black. The mean age at enrolment was 66 years and 70% of patients were male. At baseline, the mean eGFR was 44.4 mL/min/1.73 m2, with 55% of patients having an eGFR < 45 mL/min/1.73 m2, median urine albumin-to-creatinine ratio (UACR) was 853 mg/g, and mean glycated haemoglobin A1c (HbA1c) was 7.7%, 46% had a history of atherosclerotic cardiovascular disease, 30% had history of coronary artery disease, 8% had a history of cardiac failure, and the mean blood pressure was 138/76 mmHg. The mean duration of type 2 diabetes at baseline was 16.6 years and a history of diabetic retinopathy and diabetic neuropathy was reported in 47% and 26% of patients, respectively. At baseline, almost all patients were on ACEi (34%) or ARB (66%), and 97% of patients used one or more antidiabetic medications (insulin [64%], biguanides [44%], glucagon-like peptide-1 [GLP-1] receptor agonists [7%], sodium-glucose cotransporter 2 [SGLT2] inhibitors [5%]). The other most frequent medications taken at baseline were statins (74%) and calcium channel blockers (63%).
Kerendia significantly reduced the risk of the primary composite endpoint compared to placebo in a time to event analysis using the Cox proportional hazards model and log rank test (HR 0.82, 95% CI 0.73 0.92, p = 0.0009). See Figure 1, Table 6. The key secondary endpoint results (composite of CV death, non-fatal MI, non-fatal stroke, hospitalisation for heart failure) were favourable overall (HR (95% CI): 0.86 (0.75-0.99), p = 0.0344), but with an indeterminate effect observed for the 'non-fatal stroke' component with a HR (95% CI): 1.027 (0.765-1.380). Prespecified secondary time-to-event endpoints are included, see Table 6. The treatment effect for the primary and key secondary endpoints was generally consistent across subgroups, including region, eGFR, UACR, systolic blood pressure (SBP) and HbA1c at baseline. See Figure 2.
In the FIDELIO-DKD study, hyperkalaemia events were reported in 18.2% of Kerendia-treated patients compared with 9.0% of placebo-treated patients. Hospitalisation due to hyperkalaemia for the Kerendia group was 1.4% versus 0.3% in the placebo group. Hyperkalaemia leading to permanent discontinuation in patients who received Kerendia was 2.3% versus 0.9% in the placebo group.
In the FIDELIO-DKD study, glomerular filtration rate decreased events were reported in 6.4% of Kerendia-treated patients compared with 4.7% of placebo-treated patients, and those leading to permanent discontinuation in patients receiving Kerendia were 0.2% versus 0.3% in the placebo group. Patients on Kerendia experienced an initial decrease in eGFR (mean 2 mL/min/1.73 m2) that attenuated over time compared to placebo. This decrease was reversible after treatment discontinuation. The initial decrease in eGFR was associated with long term preservation of kidney function.
The FIGARO-DKD study included adults with CKD and type 2 diabetes, based on having a UACR of ≥ 30 mg/g to < 300 mg/g and an eGFR of 25 to 90 mL/min/1.73 m2, or a UACR ≥ 300 mg/g and an eGFR ≥ 60 mL/min/1.73 m2 at screening. Patients were required to have a serum potassium of ≤ 4.8 mmol/L at screening and received standard of care, including a maximum tolerated labeled dose of a RAS inhibitor (either an ACEi or ARB).
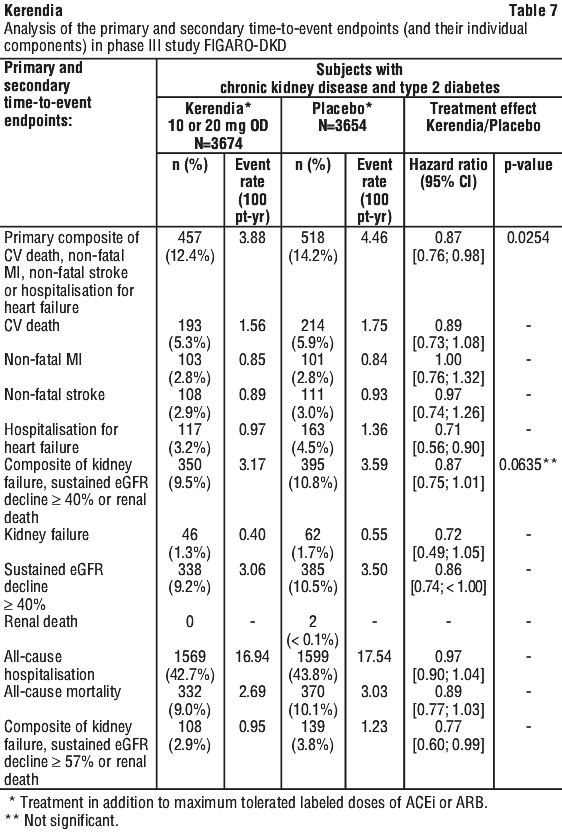
The primary endpoint in the FIGARO-DKD study was a composite of time to first occurrence of CV death, non-fatal MI, non-fatal stroke or hospitalisation for heart failure. Secondary endpoints included a composite of time to kidney failure, a sustained decline in eGFR of 40% or more compared to baseline over at least 4 weeks, or renal death and a composite of time to kidney failure, a sustained decline in eGFR of 57% or more compared to baseline, or renal death.
The trial analysed 7,328 patients randomly assigned to receive either Kerendia (N = 3674), or placebo (N = 3654) that were followed for a median duration of 3.4 years. After the end of study notification, vital status was obtained for 99.8% of patients. The trial population was 72% White, 20% Asian and 4% Black. The mean age at enrolment was 64 years and 69% of patients were male. At baseline, the mean eGFR was 67.8 mL/min/1.73 m2, with 62% of patients having an eGFR ≥ 60 mL/min/1.73 m2, median UACR was 309 mg/g, and mean glycated HbA1c was 7.7%, 45% of patients had a history of atherosclerotic cardiovascular disease, 8% had a history of cardiac failure, and the mean blood pressure was 136/77 mmHg. The mean duration of type 2 diabetes at baseline was 14.5 years and a history of diabetic retinopathy and diabetic neuropathy was reported in 31% and 28% of patients, respectively. At baseline, almost all patients were on a RAS-inhibitor and 98% of patients used one or more antidiabetic medications (insulin [54%], biguanides [69%], GLP-1 receptor agonists [8%], SGLT2 inhibitors [8%]). The other most frequent medication class taken at baseline was statins (71%).
Kerendia significantly reduced the risk of the primary composite endpoint compared to placebo in a time to event analysis using the Cox proportional hazards model and log rank test (HR 0.87, 95% CI 0.76-0.98, p = 0.0254). See Figure 3; Table 6. The treatment effect for the primary endpoint was consistent across subgroups, including region, eGFR, UACR, SBP and HbA1c at baseline. A lower incidence rate of the secondary composite outcome of kidney failure, sustained eGFR decline of 40% or more or renal death was observed in the Kerendia group compared to placebo, however this difference did not achieve statistical significance (HR 0.87, 95% CI 0.75-1.01, p = 0.0635). See Figure 4; Table 6. A lower risk of the secondary outcome of kidney failure, sustained eGFR decline of 57% or more or renal death was observed in the Kerendia group compared to placebo (HR 0.77, 95% CI 0.60-0.99). Prespecified secondary time-to-event endpoints are included in Table 7.
In the FIGARO-DKD study, hyperkalaemia events were reported in 10.7% of Kerendia-treated patients compared with 5.3% of placebo-treated patients. Hospitalisation due to hyperkalaemia for the Kerendia group was 0.6% versus < 0.1% in the placebo group. Hyperkalaemia leading to permanent discontinuation in patients who received Kerendia was 1.3% versus 0.4% in the placebo group.
In the FIGARO-DKD study, Glomerular filtration rate decreased events were reported in 4.6% of Kerendia-treated patients compared with 3.9% of placebo-treated patients, and those leading to permanent discontinuation in patients receiving Kerendia were 0.2% versus 0.1% in the placebo group. Patients on Kerendia experienced an initial decrease in eGFR of around 2 mL/min/1.73 m2 that attenuated over time compared to placebo. This decrease was reversible after treatment discontinuation. The initial decrease in eGFR was associated with long term preservation of kidney function.





Pooled analyses of FIDELIO-DKD and FIGARO-DKD.
In a pre-specified pooled analysis of the FIDELIO-DKD and FIGARO-DKD studies, finerenone reduced the risk of the CV composite endpoint of time to CV death, non-fatal MI, non-fatal stroke or hospitalisation for heart failure compared to placebo (HR 0.86 [95% CI 0.78; 0.95]). See Figure 5. The components hospitalisation for heart failure, CV death and non-fatal MI contributed to the reduction.
For the component of non-fatal stroke no treatment effect was established in the overall population. A higher proportion of patients with non-fatal stroke was observed in the subgroup of Kerendia patients without a history of CVD than in placebo (2.5% Kerendia vs 2.0% Placebo). The proportion of patients with non-fatal stroke in the subgroup of Kerendia with a history of CVD was lower compared to placebo (3.7% Kerendia vs 4.3% Placebo).
The risk of the kidney composite endpoint of time to kidney failure, a sustained decrease in eGFR of 40% or more compared to baseline or renal death was also reduced with finerenone compared to placebo (HR 0.84 [95% CI 0.77; 0.92]), as was the composite endpoint of time to kidney failure, a sustained decrease in eGFR of 57% or more compared to baseline or renal death (HR 0.76 [95% CI 0.66; 0.88]). See Figure 5.
 Heart failure. Kerendia was investigated in a randomised, double-blind, placebo-controlled, multicentre phase III study, FINEARTS-HF. The effect of Kerendia on cardiovascular outcomes was evaluated in adults with heart failure receiving either Kerendia 10 mg, 20 mg, or 40 mg once daily, or placebo.
Heart failure. Kerendia was investigated in a randomised, double-blind, placebo-controlled, multicentre phase III study, FINEARTS-HF. The effect of Kerendia on cardiovascular outcomes was evaluated in adults with heart failure receiving either Kerendia 10 mg, 20 mg, or 40 mg once daily, or placebo.
In FINEARTS-HF, patients were eligible with a diagnosis of heart failure with New York Heart Association (NYHA) class II-IV, ambulatory or hospitalised primarily for heart failure, and with documented left ventricular ejection fraction (LVEF) ≥ 40%. In addition, patients had an eGFR ≥ 25 mL/min/1.73 m2 and serum potassium ≤ 5.0 mmol/L at screening and randomization and were receiving background therapy, including diuretic treatment.
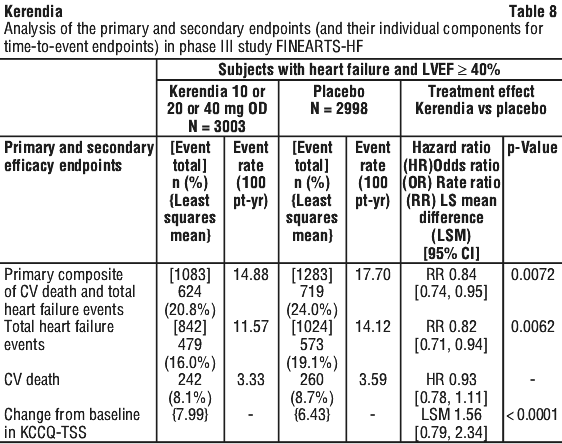
The primary endpoint was the composite of cardiovascular (CV) death and total (first and recurrent) heart failure events comprised of hospitalisation for heart failure and urgent heart failure visits. The key secondary endpoints were total (first and recurrent) heart failure events, change from baseline to Month 6, 9, and 12 in Total Symptom Score (TSS) of the Kansas City Cardiomyopathy Questionnaire (KCCQ) (which quantifies heart failure symptom frequency and severity).
The study analysed 6001 patients randomly assigned to receive either Kerendia (N = 3003) or placebo (N = 2998), with a median follow-up duration of 2.7 years. The study included 3247 (54%) patients with a heart failure event in the past 3 months, including 1219 (20%) patients randomized during the hospitalisation or within 7 days of discharge. At month 24 of the subjects treated with Kerendia, 35% were treated with 40 mg once daily, 32% with 20 mg once daily, 12% with 10 mg once daily and 1% were on a treatment interruption. At any time during treatment, approximately 80% of the patients reached their target dose. After the end of study notification, vital status was obtained for 99.7% of patients. The trial population was 79% White, 17% Asian, and 1.5% Black. The mean age at enrolment was 72 years and 46% of patients were female. At baseline, the mean LVEF was 53%, with 64% of patients having an LVEF ≥ 50%, and 69% and 30% of patients were NYHA class II and III, respectively. Mean blood pressure was 129/75 mmHg while mean body mass index (BMI) was 30 kg/m2. The median NT-proBNP was 1041 picogram/mL, the mean eGFR was 62 mL/min/1.73 m2 with 48% of patients having an eGFR < 60 mL/min/1.73 m2, and the median urinary albumin-to-creatinine ratio (UACR) was 18 mg/g. Atrial fibrillation was present for 38% of patients and 41% had diabetes mellitus. The majority of patients were on loop diuretics (87%), an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB) (79%), or an angiotensin receptor neprilysin inhibitor (ARNI) (9%), and 14% were on sodium-glucose cotransporter 2 (SGLT2) inhibitors.
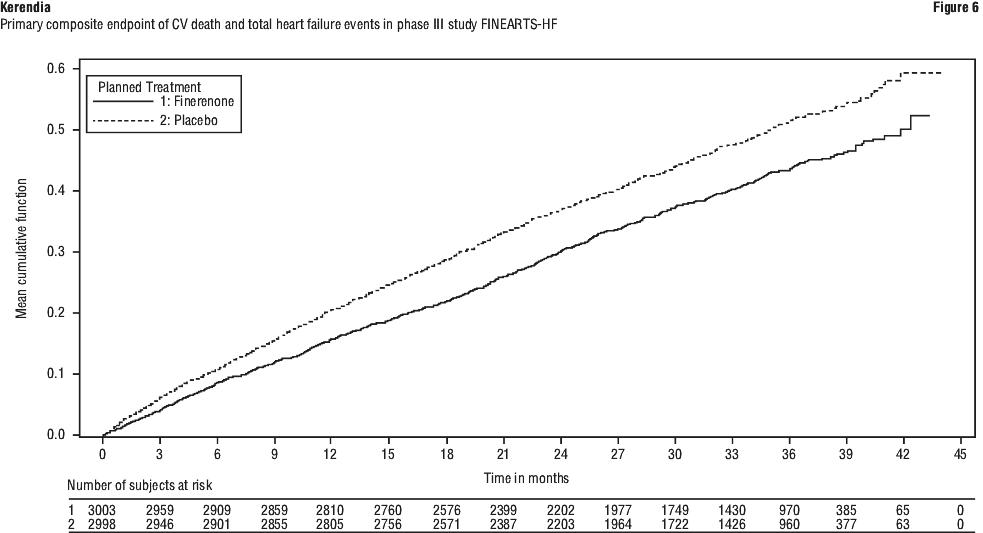
Kerendia demonstrated superiority to placebo by significantly reducing the risk of the primary composite endpoint compared to placebo (RR 0.84, 95% CI 0.74-0.95, p = 0.0072). See Table 8 and Figure 6. The effect was observed early with event curves separating from the first month and continuing to diverge throughout the study period. See Figure 6. Kerendia also demonstrated superiority to placebo on the secondary endpoints of total heart failure events (RR 0.82, 95% CI 0.71-0.94, p = 0.0062) and change from baseline to month 6, 9, and 12 in TSS of the KCCQ, indicative of improvements in symptom frequency and symptom burden (adjusted mean 1.56, 95% CI 0.79-2.34, p < 0.0001). Prespecified secondary efficacy endpoints are included in Table 8. The treatment effect for the primary and key secondary endpoints was consistent across all pre-specified subgroups, including sex, LVEF, NYHA class, eGFR, time since latest heart failure event, SGLT2 inhibitor therapy, and diabetes mellitus status.
5.2 Pharmacokinetic Properties
The concentration-effect relationship over time for UACR was characterised by a maximum effect model indicating saturation at high exposures. The model-predicted time to reach the full (99%) steady-state drug effect on UACR was 138 days. The pharmacokinetic (PK) half-life was 2-3 hours and PK steady state was achieved after 2 days, indicating timescale separation.
Absorption.
Finerenone is almost completely absorbed after oral administration. Absorption is rapid with maximum plasma concentrations (Cmax) appearing between 0.5 and 1.25 hours after tablet intake in the fasted state. The absolute bioavailability of finerenone is 43.5% due to first-pass metabolism in the gut-wall and liver. Finerenone is not a substrate of the efflux transporter P-gp in vivo. Intake with high fat, high calorie food increased finerenone AUC by up to 21%, reduced Cmax by 19% to 23% and prolonged the time to reach Cmax up to 2.5 hours. This is not clinically relevant. Therefore, finerenone can be taken with or without food (see Section 4.2 Dose and Method of Administration).
Distribution.
The volume of distribution at steady state (Vss) of finerenone is 52.6 L. The human plasma protein binding of finerenone in vitro is 91.7%, with serum albumin being the main binding protein.
Metabolism.
Approximately 90% of finerenone metabolism is mediated by CYP3A4 and 10% by CYP2C8. Four major metabolites were found in plasma, resulting from oxidation of the dihydropyridine moiety to a pyridine (M1a, M1b), subsequent hydroxylation of a methyl group (M2a) and formation of a carboxyl function (M3a). All metabolites are pharmacologically inactive.
Excretion.
The elimination of finerenone from plasma is rapid with an elimination half-life (t1/2) of about 2 to 3 hours. Excretion of unchanged finerenone represents a minor route (< 1% of dose in the urine due to glomerular filtration, < 0.2% in the faeces). About 80% of the administered dose was excreted via urine and approximately 20% of the dose was excreted via faeces, almost exclusively in the form of metabolites. With a systemic blood clearance of about 25 L/h, finerenone can be classified as a low clearance drug.
Special populations.
Patients with renal impairment.
Mild renal impairment (ClCr 60 - < 90 mL/min) did not affect finerenone AUC and Cmax. Compared to subjects with normal renal function (ClCr ≥ 90 mL/min), the effect of moderate (ClCr 30 - < 60 mL/min) or severe (ClCr < 30 mL/min) renal impairment on AUC of finerenone was similar with increases by 34-36%. Moderate or severe renal impairment had no effect on Cmax (see Section 4.2 Dose and Method of Administration).
Due to the high plasma protein binding, finerenone is not expected to be dialyzable.
Patients with hepatic impairment.
There was no change in finerenone exposure in cirrhotic subjects with mild hepatic impairment (Child-Pugh A) (see Section 4.2 Dose and Method of Administration).
In cirrhotic subjects with moderate hepatic impairment (Child-Pugh B), finerenone mean AUC was increased by 38% and Cmax was unchanged compared to healthy control subjects (see Section 4.2 Dose and Method of Administration).
There are no data in patients with severe hepatic impairment (Child-Pugh C) (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Elderly patients.
Of the 2818 patients who received Kerendia in the FIDELIO-DKD study, 58% of patients were 65 years and older, and 15% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
Of the 3671 patients who received Kerendia in the FIGARO-DKD study, 53% of patients were 65 years and older, and 14% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
Of the 3003 patients who received Kerendia in the FINEARTS study, 79% of patients were 65 years and older, and 43% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
Elderly subjects (≥ 65 years of age) exhibited higher finerenone plasma concentrations than younger subjects (≤ 45 years of age), with mean AUC and Cmax values being 34% and 51% higher in the elderly (see Section 4.2 Dose and Method of Administration). Population-pharmacokinetic analyses did not identify age as a covariate for finerenone AUC or Cmax.
Body weight.
Population-pharmacokinetic analyses identified body weight as a covariate for finerenone Cmax and AUC. Cmax and AUC of subjects with a body weight below 57 kg were estimated to be, on average, 52% and 30% higher, respectively, and of subjects with a body weight above 122 kg, 32% and 20% lower, respectively, compared to a subject between 57 and 122 kg. Dose adaptation based on body weight is not warranted (see Section 4.2 Dose and Method of Administration).
5.3 Preclinical Safety Data
Genotoxicity.
Finerenone was non-genotoxic in assays for mutagenicity in bacteria and for chromosomal aberrations in vitro (in Chinese hamster V79 cells), and the mouse bone marrow micronucleus test.
Carcinogenicity.
In 2-year carcinogenicity studies, finerenone did not increase tumour incidence in male or female rats at oral doses up to 20 and 10 mg/kg/day, or in female mice at oral doses up to 7.5 mg/kg/day (yielding exposure 19-28 or 8-11 times higher than in patients at the maximum recommended human dose of 20 or 40 mg/day, respectively, based on plasma AUC for unbound drug). In male mice, finerenone resulted in an increase in Leydig cell adenoma at 30 mg/kg/day, representing 26 or 10 times the AUCunbound in humans at the maximum recommended human dose of 20 or 40 mg/kg/day, respectively. No carcinogenicity was evident with treatment at 10 mg/kg/day, representing 7 times the AUCunbound for the 40 mg dose and 17 times the AUCunbound in humans for the 20 mg dose. Based on the known sensitivity of rodents to develop these tumours and the pharmacology-based mechanism at supratherapeutic doses as well as the margin of exposure, the increase in Leydig cell tumours observed in male mice is not considered to indicate a particular carcinogenic risk to patients treated with Kerendia.6 Pharmaceutical Particulars
6.1 List of Excipients
Tablet core.
Croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate.
Tablet coating.
Hypromellose, purified talc, titanium dioxide, iron oxide yellow (for 20 mg and 40 mg tablets), iron oxide red (for 10 mg and 40 mg tablets).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Container type: PVC/PVDC/Al blister.
Pack sizes: 14, 28, 98, 100 tablets.
Some pack sizes may not be marketed.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking it to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
1050477-31-0.
Chemical name: 4S)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1,6 naphthyridine-3-carboxamide.
Empirical formula: C21H22N4O3.
Molecular weight: 378.43 g/mol.7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Summary Table of Changes


