1 Name of Medicine
Tebentafusp.
2 Qualitative and Quantitative Composition
Each vial contains 0.1 milligrams of tebentafusp in 0.5 mL of concentrated solution for infusion.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrated solution for infusion.
Sterile, preservative-free, clear, colourless to slightly yellowish solution in a single-dose vial.
4.1 Therapeutic Indications
Kimmtrak is indicated for the treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.
4.2 Dose and Method of Administration
Kimmtrak should be administered under the supervision of a physician experienced in the use of anti-cancer agents.
Dose.
The recommended dose of Kimmtrak is:
20 micrograms on day 1;
30 micrograms on day 8;
68 micrograms on day 15;
68 micrograms once every week thereafter.
Give each dose by intravenous infusion over 15-20 minutes. Continue treatment with Kimmtrak until disease progression or unacceptable toxicity occurs.
Administer the first three doses of Kimmtrak in a healthcare setting with adequate resources (including 24-hour monitoring, medications and resuscitation equipment) to manage cytokine release syndrome (CRS). Monitor patients for CRS during infusion and for at least for 16 hours after infusion is complete (see Section 4.4 Special Warnings and Precautions for Use).
If the patient does not experience hypotension that is grade 2 or worse (requiring medical intervention) in association with the third infusion, subsequent doses can be administered in an appropriate outpatient/ambulatory care setting. Observe patients for a minimum of 30 minutes following each infusion.
Dose adjustments.
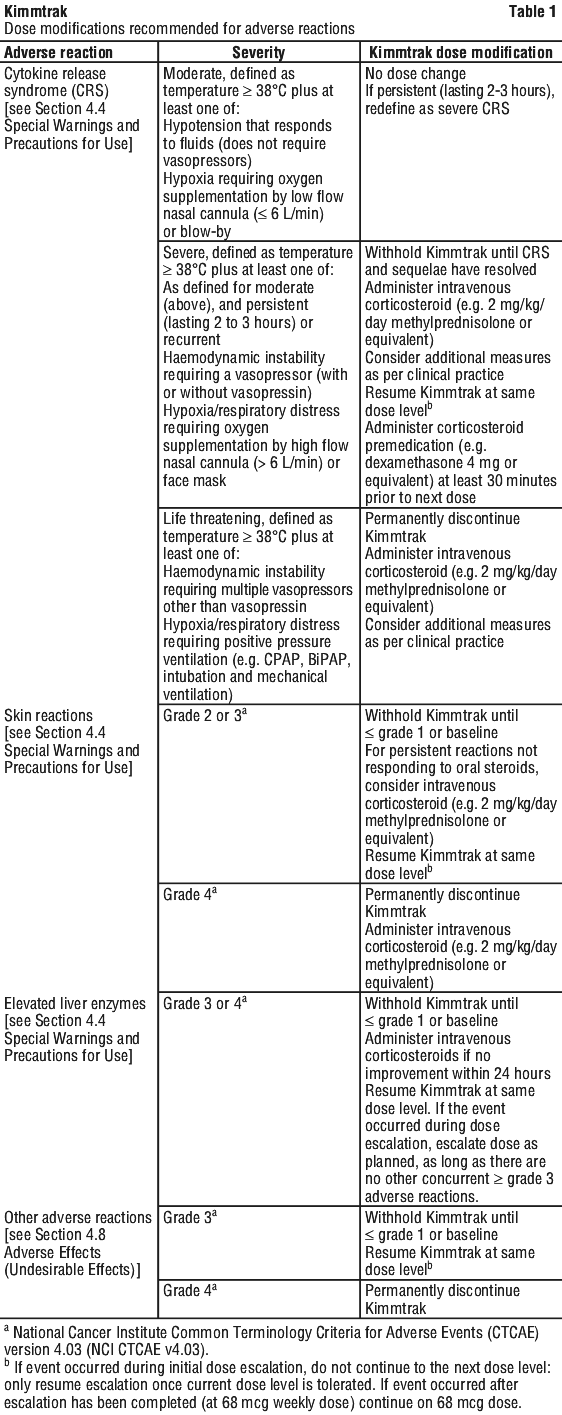
Dose modifications for Kimmtrak for adverse reactions are summarised in Table 1.
Method of administration.
Equipment. Gather the following equipment prior to preparing Kimmtrak for administration:
1 mL sterile syringes with graduations of 2 decimal places;
Sterile needles;
Locally sourced human albumin (see Table 2);
A 100 mL infusion bag containing sodium chloride 9 mg/mL (0.9%) solution for injection. The infusion bag should be constructed of polyvinyl chloride (PVC) or polyolefins (PO), such as polyethylene (PE) and polypropylene (PP);
A sterile, non-pyrogenic, low protein-binding 0.2 micron in-line filter infusion set.
Preparation. A two-step dilution process is required for preparation of the final Kimmtrak dose for infusion. Use aseptic technique for dilution and preparation of solutions for intravenous infusion. Visually inspect parenteral drug products and infusion bags for particulate matter and discoloration prior to administration, whenever solution and container permit.
Closed system transfer devices (CSTDs) must not be used for dose preparation of Kimmtrak solution for infusion.
Step 1: Prepare the infusion bag.
To prevent adsorption of tebentafusp to the infusion bag and other components of the drug delivery system, prepare a solution of 250 microgram/mL human albumin in 0.9% sodium chloride as follows:
1a. Using a 1 mL sterile syringe with graduations of 2 decimal places and a sterile needle, withdraw the calculated volume of source human albumin into the syringe (see Table 2 for calculation examples) and add to the 100 mL bag containing 0.9% sodium chloride solution for injection, to achieve a final human albumin concentration of 250 microgram/mL. Do not shake the infusion bag.
 1b. Gently homogenise the 250 microgram/mL human albumin solution by completing the following steps:
1b. Gently homogenise the 250 microgram/mL human albumin solution by completing the following steps:
i. Gently invert the infusion bag so that the bag is upside down, with the entry port positioned at the top. Then tap the side of the port tubing to ensure that any residual concentrated albumin solution is released into the bulk solution.
ii. Gently rotate the bag lengthwise (360 degrees: from upside-down to right-side-up, and back again) at least 5 times.
iii. Repeat (i) and (ii) an additional three times.
Step 2: Preparation of Kimmtrak solution for infusion.
Do not shake the Kimmtrak vial.
2a. Using a 1 mL syringe with graduations of 2 decimal places and a sterile needle, withdraw the required volume of Kimmtrak 200 micrograms/mL as per the dose required (see Table 3) and add to the prepared 100 mL infusion bag containing sodium chloride 9 mg/mL (0.9%) solution for injection plus human albumin (250 microgram/mL). Do not flush the needle and syringe on transfer.
 2b. Each vial of Kimmtrak is for single use in one patient only. Discard any residue. Discard the vial containing the unused portion of Kimmtrak in accordance with local requirements.
2b. Each vial of Kimmtrak is for single use in one patient only. Discard any residue. Discard the vial containing the unused portion of Kimmtrak in accordance with local requirements.
2c. Gently mix the infusion bag by following the same procedure outlined in step 1b.
Administration. Immediately administer the diluted solution by intravenous infusion over 15-20 minutes through a dedicated intravenous line, using a sterile, non-pyrogenic, low protein binding 0.2 micron in-line filter infusion set. Administer the entire contents of the Kimmtrak infusion bag to the patient.
Kimmtrak does not contain a preservative. The infusion should be completed within 4 hours from the time of preparation. During the 4-hour window, the Kimmtrak infusion bag should remain below 30°C.
Upon completion of Kimmtrak infusion, flush the infusion line with an adequate volume of sterile sodium chloride 9 mg/mL (0.9%) solution for injection to ensure that the entire contents of the Kimmtrak infusion bag are administered.
Do not administer Kimmtrak as an intravenous push or bolus, or by any non-intravenous route of administration. Do not mix Kimmtrak with other drugs or administer other drugs through the same intravenous line.
Storage of diluted solution for infusion. If not used immediately, store diluted solution for infusion in a refrigerator at 2°C to 8°C and infuse within 24 hours from the time of preparation. The 24-hour window includes refrigerator storage time, the time taken for the infusion bag to re-equilibrate to room temperature (below 30°C) and the duration of the infusion.
Once removed from the refrigerator, do not refrigerate the Kimmtrak infusion bag again. Do not freeze. Discard unused Kimmtrak solution beyond the recommended storage time.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Traceability.
To improve the traceability of biological medicinal products, the name and the batch number of the administered product should be clearly recorded.
Cytokine release syndrome (CRS).
CRS, which can be life threatening, occurred in patients receiving Kimmtrak (see Section 4.8 Adverse Effects (Undesirable Effects), Description of selected adverse reactions).
Manifestations of CRS may include fever, hypotension, hypoxia, chills, nausea, vomiting, rash, elevated transaminases, fatigue and headache. CRS has been associated with organ dysfunction, including hepatic, renal, pancreatic, cardiac, and pulmonary dysfunction.
Before initiating Kimmtrak infusion, ensure patients are euvolemic and that healthcare providers with adequate expertise and immediate access to medications and resuscitation equipment are available to manage CRS. For patients with pre-existing adrenal insufficiency on maintenance systemic corticosteroids, consider adjusting the corticosteroid dose to manage the risk of hypotension.
Closely monitor patients for signs or symptoms of CRS during and after infusions of Kimmtrak (see Section 4.2 Dose and Method of Administration). Monitor fluid status, vital signs and oxygenation level and provide appropriate therapy. Withhold or discontinue Kimmtrak depending on persistence and severity of CRS (see Section 4.2 Dose and Method of Administration, Table 1).
Skin reactions.
Skin reactions occurred in patients receiving Kimmtrak, including rash, pruritus, erythema and cutaneous oedema (see Section 4.8 Adverse Effects (Undesirable Effects), Description of selected adverse reactions).
Treat skin reactions with antihistamines and topical or systemic steroids based on persistence or severity of symptoms. Withhold or discontinue Kimmtrak depending on persistence and severity of skin reactions (see Section 4.2 Dose and Method of Administration, Table 1).
Elevated hepatic enzymes.
Elevation of hepatic enzymes occurred in patients receiving Kimmtrak (see Section 4.8 Adverse Effects (Undesirable Effects), Description of selected adverse reactions).
Monitor alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total blood bilirubin before and during treatment with Kimmtrak. Withhold Kimmtrak according to severity (see Section 4.2 Dose and Method of Administration, Table 1).
Use in hepatic impairment.
Dose adjustment is not necessary in patients with mild hepatic impairment. Tebentafusp has not been studied in patient with moderate to severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Use in renal impairment.
Dose adjustment is not necessary in patients with mild to moderate renal dysfunction. Tebentafusp has not been studied in patient with severe renal impairment (see Section 5.2 Pharmacokinetic Properties).
Use in the elderly.
Dose adjustment is not necessary for elderly patients (≥ 65 years of age).
Of the 245 patients with metastatic uveal melanoma treated with Kimmtrak in study IMCgp100-202, 47% were 65 years of age or older and 9% were 75 years of age or older. No meaningful differences in safety or efficacy were observed between patients ≥ 65 years of age compared to younger adult patients.
Paediatric use.
The safety and efficacy of Kimmtrak in children under the age of 18 years has not been established.
Effects on laboratory tests.
No data are available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No formal drug interaction studies have been performed with tebentafusp.
Kimmtrak treatment causes transient release of cytokines that may suppress CYP450 enzymes. The effect attenuates with repeat dosing of Kimmtrak, such that cytokine levels are highest during the 24-hour period after each of the first three doses. In patients who are receiving concomitant CYP450 substrates, particularly those with a narrow therapeutic index, consider the possibility of interaction and monitor for toxicity (e.g. warfarin) or drug concentrations (e.g. cyclosporine). Adjust the dose of the concomitant drug as needed.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No fertility studies have been conducted with tebentafusp. There are no data on the effect of tebentafusp on human fertility.
(Category C)
Based on the mechanism of action, Kimmtrak may cause foetal harm when administered to a pregnant person (see Section 5.1 Pharmacodynamic Properties). There are no data regarding the use of tebentafusp in pregnancy. No animal reproduction studies or developmental toxicity studies have been conducted with tebentfusp. Molecules of similar molecular weight can cross the placenta resulting in foetal exposure. Advise patients of the potential risk to a foetus.
Patients of childbearing potential should use effective contraception during treatment with tebentafusp and for at least 1 week after last dose of tebentafusp.
Verify pregnancy status in patients of reproductive potential prior to initiating tebentafusp treatment.
There is no information regarding the presence of tebentafusp in human milk, the effect on the breastfed child, or the effects on milk production. Because tebentafusp may be excreted in human milk and because of the potential for serious adverse reactions in a breastfed child, advise patients not to breastfeed during treatment with Kimmtrak and for at least 1 week after the last dose.4.7 Effects on Ability to Drive and Use Machines
Tebentafusp has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Summary of safety profile.
The safety of Kimmtrak was evaluated in study IMCgp100-202, a randomised (2:1), open-label, active-controlled trial in patients who had not received prior systemic therapy for metastatic or advanced uveal melanoma (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Patients received either Kimmtrak administered at 20 microgram intravenously on day 1, 30 microgram intravenously on day 8, 68 microgram intravenously on day 15, and 68 microgram intravenously once every week thereafter (n = 245) or investigator's choice treatment (n = 111). The median duration of exposure was 5.3 months (range: 0.3 to 33 months) in patients treated with Kimmtrak.
Serious adverse events occurred in 28% of patients who received Kimmtrak. Serious adverse events that occurred in > 2% of patients were cytokine release syndrome (10%), rashes (4.5%), pyrexia (2.4%), and hypotension (2%). One patient (0.4%) experienced a fatal adverse event (pulmonary embolism).
Adverse events led to permanent discontinuation in 3.3% of patients who received Kimmtrak. Adverse events that led to permanent discontinuation of Kimmtrak were anaphylactic reaction, brain oedema, cytokine release syndrome, fatigue, hepatotoxicity, hypotension, and nausea (each 0.4%).
Adverse events resulting in dose interruption occurred in 25% of patients who received Kimmtrak. Adverse events which required dose interruption in ≥ 2% of patients included fatigue (3.7%), lipase increased (2.9%), pyrexia (2.4%), alanine aminotransferase increase (2%), and aspartate aminotransferase increase (2%).
Adverse events leading to dose reduction occurred in 5% of patients who received Kimmtrak. Adverse events which required dose reduction in ≥ 2% of patients were cytokine release syndrome (2.4%), and rashes (2%).
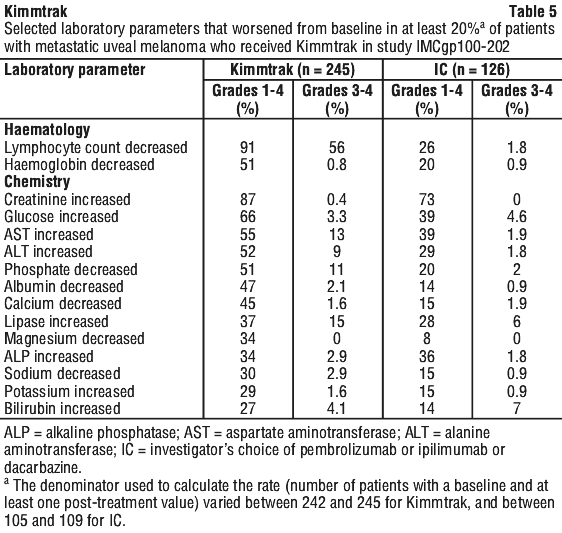
The most common adverse events (≥ 30%) in patients who received Kimmtrak (see Table 4) were cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, oedema, hypotension, dry skin, headache and vomiting. The most common (≥ 50%) laboratory abnormalities in patients who received Kimmtrak were decreased lymphocyte count, increased creatinine, increased glucose, increased AST, increased ALT, decreased haemoglobin, and decreased phosphate.
Tabulated list of adverse reactions.
Table 4 summarises the adverse events observed in study IMCgp100-202.
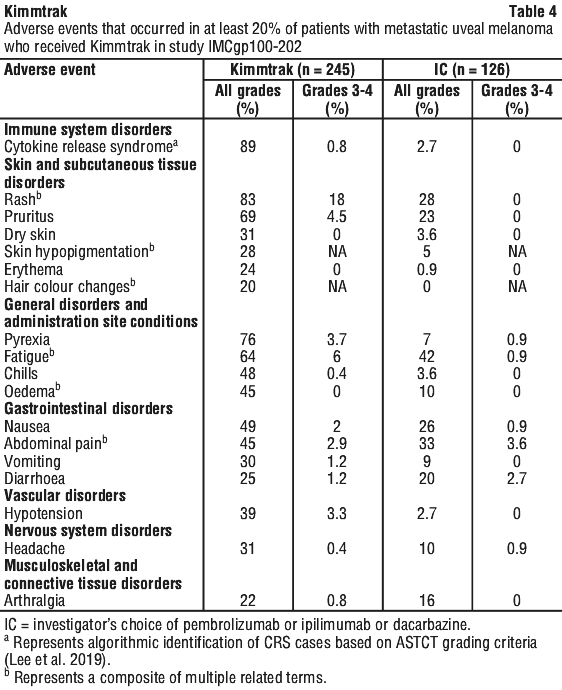 Clinically relevant adverse reactions that occurred in < 20% of patients who received Kimmtrak included back pain, decreased appetite, constipation, hypertension, tachycardia or sinus tachycardia, dyspnoea, paraesthesia, dizziness, flushing, muscle spasms, myalgia, pain in extremity, alopecia, skin hyperpigmentation, influenza-like illness, oropharyngeal pain and night sweats.
Clinically relevant adverse reactions that occurred in < 20% of patients who received Kimmtrak included back pain, decreased appetite, constipation, hypertension, tachycardia or sinus tachycardia, dyspnoea, paraesthesia, dizziness, flushing, muscle spasms, myalgia, pain in extremity, alopecia, skin hyperpigmentation, influenza-like illness, oropharyngeal pain and night sweats.
Table 5 summarises selected laboratory abnormalities observed in study IMCgp100-202.
Description of selected adverse reactions.
Cytokine release syndrome (CRS).
In study IMCgp100-202, CRS of at least grade 2 severity occurred with Kimmtrak infusion at least once for 77% of patients, and 60% experienced it at least twice. In most cases, CRS started on the day of infusion. Fever was noted in nearly all cases of CRS, generally within the first 8 hours after Kimmtrak infusion.
Systemic corticosteroids were received by 23% of patients, supplemental oxygen was received by 8% of patients, and vasopressor treatment was received by 0.8% of patients to treat CRS during at least one infusion. Among cases that resolved, the median time to resolution of CRS was 2 days. CRS led to permanent treatment discontinuation in 1.2% of patients.
Acute skin reactions.
In study IMCgp100-202, acute skin reactions occurred in 91% of patients who received Kimmtrak, including rash (grouped term; 83%), pruritus (grouped term; 69%), erythema (25%) and cutaneous oedema (grouped term; 27%). Skin reactions were all grade 1 (28%), grade 2 (44%), or grade 3 (21%).
The frequency of acute skin reactions decreased after the first three Kimmtrak infusions. The rate of grade 3 reactions was 17% following dose 1; 10% following dose 2; 8% following dose 3; and 3% following dose 4. The median time to onset of acute skin reactions in patients receiving Kimmtrak was 1 day and median time to improvement to ≤ grade 1 was 6 days. There were no permanent discontinuations of Kimmtrak due to acute skin reactions.
Elevated hepatic enzymes.
In study IMCgp100-202, 95% of patients had liver metastasis at enrolment, and ALT/AST increases to ≥ grade 1 were observed in 65% of patients who received Kimmtrak. No deaths due to ALT/AST elevations were observed and more than 90% of patients were able to continue treatment beyond worst grade ALT/AST elevation. Most patients who experienced ALT/AST elevations (73%) did so within the first 3 infusions of Kimmtrak. Most patients (56%) who experienced grade 3 or 4 ALT/AST elevations had improvement to ≤ grade 1 within 7 days.
Elevations in bilirubin were reported in 27% of patients receiving Kimmtrak. In 58% of these cases, an increase in size of liver metastasis was seen at the tumour assessment that occurred closest to the date of bilirubin elevation (tumour assessments were scheduled 12 weekly).
Immunogenicity.
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralising antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Treatment-emergent anti-drug antibodies (ADA) against tebentafusp were detected in 33% and 29% of patients receiving tebentafusp across all doses in study IMCgp100-102 and study IMCgp100-202, respectively. The median onset time to ADA formation was 6-9 weeks after tebentafusp treatment. The ability of these binding ADA to neutralise tebentafusp is unknown. Tebentafusp clearance increased in patients with high titer ADAs (see Section 5.2 Pharmacokinetic Properties). Exploratory analyses with limited data were not suggestive of a relationship between the formation of ADA and the frequency or severity of hypersensitivity-related adverse reactions or decreased overall survival.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no information on overdose with tebentafusp. In case of overdose, patients should be closely monitored for signs or symptoms of adverse reactions and appropriate symptomatic treatment should be instituted immediately.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: antineoplastic agents; other antineoplastic agents, ATC code: L01XX75.
Mechanism of action.
Tebentafusp is a bispecific fusion protein, comprised of a T cell receptor (TCR) fused to an antibody fragment with specificity for the CD3 (cluster of differentiation 3) receptor, found on polyclonal T cells. The TCR has specificity for a gp100 peptide (expressed preferentially in melanoma cells) presented by human leukocyte antigen-A*02:01 (HLA-A*02:01).
In vitro, tebentafusp bound to HLA-A*02:01-positive uveal melanoma cells and activated polyclonal T cells to release inflammatory cytokines and cytolytic proteins, which results in direct lysis of uveal melanoma tumour cells.
Pharmacodynamic effects.
Lymphocyte counts declined the day after each of the first three tebentafusp doses and returned to baseline prior to subsequent doses.
Serum levels of cytokines (IFN-γ, TNFα, IL-2, IL-6, IL-10 and IL-1RA) and chemokines (CXCL9, CXCL10, CXCL11, hepatocyte growth factor, and monocyte chemoattractant protein-1) were increased after each of the first three tebentafusp doses, peaking 8 to 24 hours after treatment and returning to baseline prior to subsequent doses. The intensity of cytokine elevation, and the proportion of patients in whom it occurred, decreased in subsequent treatment cycles.
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of Kimmtrak have not been fully characterised.
Clinical trials.
Study IMCgp100-202 was a randomised, open-label, multicentre trial that enrolled patients with metastatic uveal melanoma who had a HLA-A*02:01 genotype according to a centrally-conducted clinical trial assay. Patients were excluded if they received previous systemic treatment or localised (liver-directed) therapy for metastatic uveal melanoma, however, prior surgical resection of oligometastatic disease was permitted. Patients with clinically significant cardiac disease and symptomatic or untreated brain metastasis were also excluded.
Patients were randomised (2:1) to either receive Kimmtrak by intravenous infusion at a dose of 20 microgram on day 1, 30 microgram on day 8, 68 microgram on day 15, and 68 microgram once every week thereafter (n = 252) or to receive investigator's choice (n = 126) of pembrolizumab, ipilimumab, or dacarbazine. Randomisation was stratified by lactate dehydrogenase (LDH) level at study entry. Across both arms, treatment was ceased on disease progression (unless the patient was otherwise deriving benefit), or for unacceptable toxicity.
The primary efficacy outcome was overall survival (OS). Additional efficacy outcomes were investigator-assessed progression free survival (PFS) and objective response rate (ORR) per RECIST v1.1.
The median age was 64 years (range 23 to 92 years); 87% of patients were White and 50% were female. Baseline ECOG performance status was 0 (73%) or 1 (21%) or 2 (0.3%); 36% had an elevated LDH level, and 94% had liver metastasis.
The efficacy results are summarised in Table 6 and Figure 1.
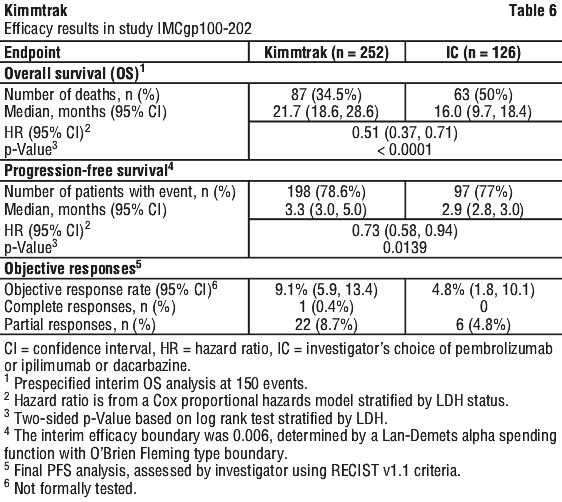
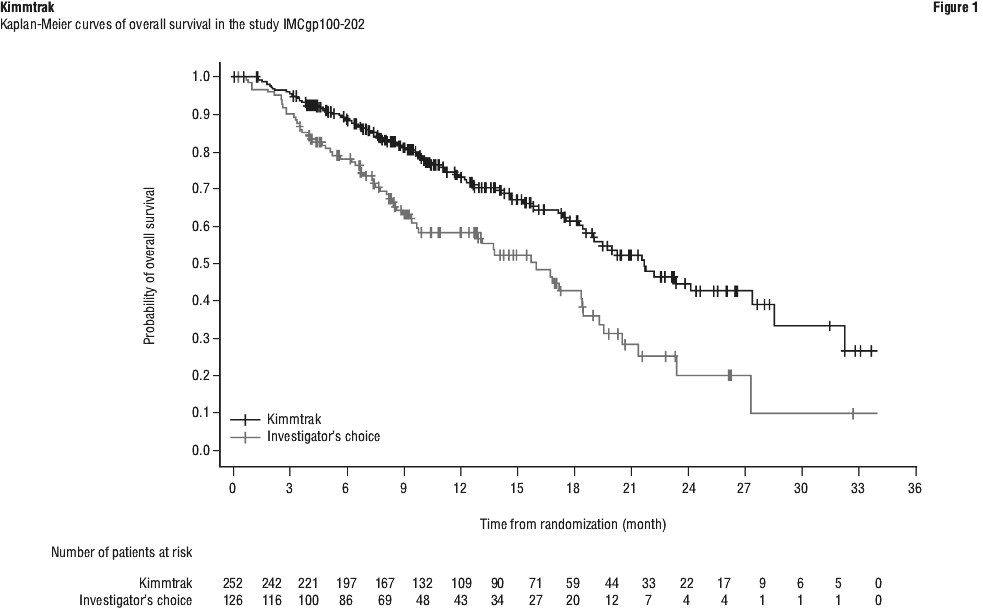 In the pre-specified subgroup analysis by LDH status, the HR for overall survival was 0.35 (95% CI: 0.21, 0.60) in the LDH ≤ ULN and 0.70 (95% CI: 0.46, 1.09) in the LDH ≥ ULN subgroup. In patients with ≤ 3 cm liver lesion diameter, treatment with tebentafusp showed improved OS compared to the investigator's choice treated group [HR = 0.36 (95% CI: 0.21, 0.61)].
In the pre-specified subgroup analysis by LDH status, the HR for overall survival was 0.35 (95% CI: 0.21, 0.60) in the LDH ≤ ULN and 0.70 (95% CI: 0.46, 1.09) in the LDH ≥ ULN subgroup. In patients with ≤ 3 cm liver lesion diameter, treatment with tebentafusp showed improved OS compared to the investigator's choice treated group [HR = 0.36 (95% CI: 0.21, 0.61)].
5.2 Pharmacokinetic Properties
Absorption.
After a single dose administration, tebentafusp Cmax and AUC0-7d increased in an approximately dose-proportional manner, over a dose range of 20 microgram to 68 microgram (0.3 to 1 times the recommended dose). Following administration of tebentafusp at the recommended dose in patients with metastatic uveal melanoma, the steady-state geometric mean (% CV) Cmax was 13 nanogram/mL (35%) and AUC0-7d was 4.6 nanogram.day/mL (23%), with no accumulation.
Distribution.
The geometric mean (% CV) steady-state volume of distribution for tebentafusp is 7.56 L (24%).
Metabolism.
As a protein, tebentafusp is expected to be catabolised into small peptides and amino acids.
Excretion.
The geometric mean (% CV) clearance of tebentafusp is 16.4 L/d (25%) and the median terminal half-life is 7.5 hours (range: 6.8-7.5 hours).
Special populations.
Weight (43 to 163 kg), sex (48% female), age (23 to 91 years), mild to moderate renal impairment (creatinine clearance estimated by C-G formula [CLcr] of 30 to 89 mL/min), and mild hepatic impairment (elevated aspartate aminotransferase [AST] with normal total bilirubin [TB], or TB > 1 to 1.5 times the upper limit of normal [ULN] with any AST) had no clinically significant effect on the pharmacokinetics of tebentafusp.
Tebentafusp has not been studied in patients with severe renal impairment (CLcr < 30 mL/min) or in patients with moderate hepatic impairment (TB > 1.5 to 3 x ULN, any AST) or severe hepatic impairment (TB > 3 to 10 x ULN, any AST).
Immunogenicity.
Median titer in the ADA-positive subgroup was 8192 across the 67 treatment cycles. The exposure (AUC0-7 days) of tebentafusp decreased by 97% and terminal half-life decreased to 10-14 minutes in patients with ADA titers greater than 8192.
5.3 Preclinical Safety Data
Genotoxicity.
No genotoxicity studies have been conducted with tebentafusp.
Carcinogenicity.
No carcinogenicity studies have been conducted with tebentafusp.6 Pharmaceutical Particulars
6.1 List of Excipients
Citric acid monohydrate, dibasic sodium phosphate, mannitol, trehalose dihydrate, polysorbate 20, water for injection.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in Section 4.2 Dose and Method of Administration.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
For the in-use shelf life and storage conditions after vial opening and after dilution of the medicinal product, see Section 4.2 Dose and Method of Administration.
6.4 Special Precautions for Storage
Store at 2°C to 8°C. (Refrigerate. Do not freeze). Keep the vial in the outer carton in order to protect from light.
6.5 Nature and Contents of Container
Type I glass vial with a bromobutyl rubber stopper and an aluminium/plastic flip-off seal, containing 0.5 mL concentrated solution for infusion.
Pack size of 1 vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
Tebentafusp is a bispecific gp100-targeted T cell receptor fusion protein with an approximate molecular weight of 77 kDa. Tebentafusp is produced by recombinant DNA technology in Escherichia coli cells.
CAS number.
1874157-95-5.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
