What is in this leaflet
Please read this leaflet carefully before you start using KISQALI.
This leaflet answers some common questions about KISQALI.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine.
You can also download the most up to date leaflet from www.novartis.com.au or from www.ebs.tga.gov.au/. Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking KISQALI against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What KISQALI is used for
KISQALI tablets contain an active substance called ribociclib. It is used to treat hormone receptor (HR)- positive, human epidermal growth factor receptor 2 (HER2) - negative breast cancer that is locally advanced or may have also spread to other parts of the body (metastatic).
KISQALI is used in combination with a second medicine (either an aromatase inhibitor or fulvestrant), which are used as hormonal anticancer therapies. Your doctor or pharmacist will advise you as to which medication you are taking with KISQALI.
If KISQALI is used in women who have not reached menopause in combination with an aromatase inhibitor, a third medicine must also be used from the group of luteinising hormone-releasing hormone (LHRH) agonists. This medicine controls the function of your ovaries, by reducing the amount of oestrogen (a hormone) that is produced by your body.
KISQALI is believed to work by blocking the effects of types of enzymes, called cyclin dependent kinases (CDK) that chemically signal cancer cells to grow and multiply. By blocking these enzymes, KISQALI may delay the growth of breast cancer cells.
Ask your doctor or pharmacist if you have any questions about why this medicine has been prescribed for you.
Your doctor may have prescribed KISQALI for another reason.
This medicine should not be used in children and adolescents, under the age of 18 years.
This medicine is available only with a doctor's prescription. It is not addictive.
Please also read the Consumer Medicine Information (CMI) for the other medicines used in combination with KISQALI carefully as well. That is, the:
- Aromatase inhibitor OR fulvestrant, depending on what your doctor has prescribed.
- LHRH agonist (if you are a premenopausal or perimenopausal woman).
Before you take KISQALI
When you must not use it
Do not take KISQALI if you have an allergy to:
- ribociclib
- any cyclin dependent kinase inhibitors
- soya lecithin
- any of the other ingredients of KISQALI tablets (see list at the end of this leaflet).
Some of the symptoms of an allergic reaction may include:
- Severe itching of the skin, with a red rash, or raised bumps;
- Swelling of the face, lips, tongue, throat, or other parts of the body;
- Difficulty in breathing or swallowing;
- Dizziness.
If you think you may be allergic, ask your doctor for advice.
If you experience an allergic reaction, stop using the medicine and tell your doctor or pharmacist immediately.
Do not take KISQALI if you have:
- A heart problem known as QT prolongation
This is caused by a change in the electrical activity of the heart, and is seen by your doctor on an ECG (electrocardiogram), or - Conditions which put you at risk of getting QT prolongation
Such as:
- slow heartbeat
- low potassium, magnesium, calcium or phosphorous levels in your blood
- family history of QT prolongation, or - Taking other medicines which prolong the QT interval
Your doctor or pharmacist will know more about this.
Women of childbearing age who recently became postmenopausal or peri menopausal should not commence treatment with KISQALI until your post-menopausal status is fully established.
Do not take KISQALI if you are pregnant or breastfeeding. It may affect your baby if you take it while you are pregnant or breastfeeding.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. The expiry date refers to the last day of that month.
If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you are allergic to any other medicines, foods, dyes or preservatives. Your doctor will want to know if you are prone to allergies.
Tell your doctor or pharmacist before taking KISQALI if you have any of the following conditions:
- Fever, sore throat or mouth ulcers due to infections (signs of low level of white blood cells)
- Any problems with your liver or previously had any type of liver disease
- Have or had heart failure, a heart attack, heart disorders or heart rhythm disorders, such as an irregular heartbeat, including a condition called prolonged QT syndrome (QT interval prolongation)
- Low levels of potassium, magnesium, calcium, or phosphorous in your blood
- Are still having periods
- Are pregnant, think you may be pregnant, or plan to become pregnant
- Plan to breastfeed
- Are taking any medicines or supplements (see "Taking other medicines").
Taking other medicines
Before you take KISQALI, tell your doctor or pharmacist if you are taking or have recently taken any other medicines, including medicines or supplements that you can buy without a prescription from a pharmacy, supermarket or health food shop.
These may interact with KISQALI. In particular, some medicines used to treat:
- Fungal infections, such as ketoconazole, itraconazole, voriconazole or posaconazole;
- Certain types of bacterial infections, such as erythromycin, clarithromycin, azithromycin, moxifloxacin, levofloxacin, norfloxacin, and ciprofloxacin
- HIV/AIDS such as: ritonavir, saquinavir, idinavir, lopinavir, nelfinavir, telaprevir and efavirenz;
- Seizures or fits (anti-epileptics) such as carbamazepine, phenytoin, rifampin and midazolam;
- Heart rhythm problems such as amiodarone, disopyramide, procainamide, quinidine and sotalol;
- Depression, anxiety, sleep problems, or other conditions with a herbal product called St John's Wort (also known as Hypericum perforatum).
Ask your doctor or pharmacist if you are not sure whether your medicine is one of the medicines listed above.
Keep a list of the medicines you take, so you can show it to your doctor, nurse or pharmacist when you get a new medicine.
Other medicines may be affected by KISQALI or they may affect how well KISQALI works. This includes medicines or supplements obtained without a prescription and/or herbal medicines.
You should also tell your doctor if you are already taking KISQALI if you are prescribed a new medicine that you have not taken previously during KISQALI treatment.
Your doctor or pharmacist can tell you what to do when taking KISQALI with other medicines.
Women of child-bearing potential
If you are pregnant or breast-feeding, think you might be pregnant, or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
KISQALI may harm your unborn baby if you are pregnant.
KISQALI may harm your baby if you are breastfeeding. Your doctor will discuss with you the potential risks of taking KISQALI during pregnancy or when breastfeeding.
If until recently you still had menstrual periods, you should delay commencing KISQALI until your post-menopausal status is determined.
If you are able to become pregnant, your doctor will discuss with you the potential risks of taking KISQALI during pregnancy or breastfeeding.
Women who are able to become pregnant should have a negative pregnancy test result before starting treatment and use effective birth control during treatment and for at least 21 days after stopping KISQALI.
Men taking KISQALI
KISQALI may reduce fertility in male patients.
How to take KISQALI
KISQALI is taken in repeating cycles of 28 days (4 weeks).
KISQALI is taken each day for 21 days, followed by a treatment break of 7 days when KISQALI tablets are not taken.
Always take KISQALI exactly as your doctor and pharmacist has told you. Your doctor or pharmacist will tell you exactly how many tablets to take and which days to take them on. These directions may differ from the information contained in this leaflet.
Check with your doctor or pharmacist if you are not sure.
Do not change the KISQALI dose or schedule without talking to your doctor.
If you do not understand the instructions on the pack, ask your doctor or pharmacist for help.
How much KISQALI to take
DAYS 1 to 21 (of repeating 28 day cycles)
- The usual starting dose of KISQALI is 600 mg (three KISQALI 200 mg tablets) once daily.
- Your doctor will tell you exactly how many tablets of KISQALI to take.
Do not exceed the recommended dose prescribed by your doctor. It is very important to follow your doctor’s recommendations. If you have certain side effects, your doctor may ask you to change to a lower dose, or ask you to skip the dose or to discontinue treatment.
DAYS 22 to 28 (of repeating 28 day cycles)
Do not take any KISQALI tablets this week.
It is very important that you do not take KISQALI during Week 4 of the cycle. That is on DAYS 22 to 28 of every cycle.
The 7 day break when you do not take KISQALI tablets will help your body to recover and decrease the risks of getting any potentially serious side effects or an infection.
When taking KISQALI and an aromatase inhibitor in combination, keep taking the aromatase inhibitor ONLY on these 7 days, as directed by your doctor.
Resume taking KISQALI once a day on the following week.
If you are aged 65 years and over, your dose does not need to be modified.
When to take it
Take KISQALI tablets at about the same time each day, preferably in the morning on days 1 to 21 of a 28 day cycle. Taking the tablets at the same time each day will have the best effect. It will also help you remember when to take them.
KISQALI tablet packs have a fifth flap to help you keep track of your doses during each treatment cycle.
Write in the days of the week starting with the first day of your treatment. Cross off a circle after each tablet that you take, in each week of the cycle, as shown in the pack example.
How to take it
Swallow KISQALI tablets whole with a glass of water or other liquid.
Do not chew, crush, or split the tablets prior to swallowing. No tablet should be ingested if it is broken, cracked, or otherwise not intact.
Taking the aromatase inhibitor or fulvestrant in combination with KISQALI
These other medicines are supplied separately.
Your doctor will tell you the dose of the aromatase inhibitor or fulvestrant, or LHRH agonist that you should take and when you should take it.
Keep taking theses medicines as directed by your doctor.
Taking with food or drinks
KISQALI tablets can be taken with or without food.
However, do not eat grapefruit (or drink grapefruit juice), pomelos, star-fruit, or Seville oranges during your treatment with KISQALI.
It may change the way that KISQALI is absorbed in your body.
How long to take it
This is a long-term treatment, which may continue for many months or years.
Continue taking KISQALI once a day on days 1 to 21, of repeating 28 day cycles, for as long as your doctor tells you to do so.
Your doctor will regularly check your condition to ensure that KISQALI treatment is having the desired effect.
If you are unsure, talk to your doctor.
If you forget to take it
If you miss a dose of KISQALI on Days 1 to 21, skip the missed dose that day. Take your next dose at your regular time on the following day.
Do not take a double dose to make up for a forgotten or a missed dose. Instead, wait until it is time for your next dose and then take your usual dose.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take or skip KISQALI, keep a treatment diary, or ask your doctor, nurse or pharmacist for some hints.
If you take more KISQALI than you should (overdose)
If you think that you have accidentally taken too many tablets or that someone else may have accidentally taken your medicine (or the aromatase inhibitor), immediately telephone your doctor, a hospital, or the Poisons Information Centre (telephone 13 11 26) for advice. Go to Accident and Emergency at your nearest hospital. Take the KISQALI pack with you.
Do this even if there are no signs of discomfort or poisoning. You may need medical attention.
Keep the telephone numbers for these places handy.
While you are taking KISQALI
Things you must do
If you become pregnant while taking KISQALI, tell your doctor immediately. You should not take this medicine while you are pregnant.
Follow your doctor's instructions carefully. If you do not follow your doctor's instructions, your treatment may not help or you may have unwanted side effects.
Be sure to keep all of your doctor's appointments so that your progress can be checked. You will have regular blood tests before and during treatment with KISQALI to monitor your liver function, the amount of blood cells, and electrolytes (blood salts including potassium, calcium, magnesium and phosphate) in your body.
The electrical activity of your heart will also be checked before and during treatment (with a test called an electrocardiogram or ECG). These tests can be affected by KISQALI.
Your doctor will also check your lung function.
If necessary, your doctor may decide to temporarily stop or reduce your KISQALI dose to allow your liver function, blood cells, electrolytes, heart activity and lungs to recover. Your doctor may also decide to stop treatment permanently.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking KISQALI.
Tell any other doctor, dentist or pharmacist who treats you that you are taking KISQALI.
Be careful when driving or using machines during your treatment with KISQALI.
Treatment with KISQALI may lead to tiredness, dizziness or vertigo.
Things you must not do
Do not take KISQALI to treat any other complaints unless your doctor says you can.
Do not give this medicine to anyone else, even if their symptoms seem to be similar to yours.
Things to be careful of
When taking KISQALI with an aromatase inhibitor, do not skip the aromatase inhibitor on any day. It must be taken EVERY day as directed in the 28 day cycle.
Side effects
Like all medicines, KISQALI can cause unwanted side effects, in addition to its beneficial effects. Sometimes they are serious, and you may need medical treatment.
The side effects that you might experience with KISQALI are different to what you may have experienced in the past, while taking hormonal therapy only.
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking KISQALI.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
SERIOUS SIDE EFFECTS
Stop taking KISQALI and tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following symptoms during treatment with KISQALI:
- Signs of an allergic reaction (as given on page 1)
- Fever, sweats or chills, cough, flu like symptoms, weight loss, shortness of breath, blood in your phlegm, sores on your body
- warm or painful areas on your body, diarrhoea or stomach pain, feeling very tired (these may be signs of infections)
- Fever, sore throat or mouth ulcers due to infections
- Tiredness, itchy yellow skin or yellowing of the whites of your eyes, nausea or vomiting, loss of appetite, pain in the upper right side of the belly (abdomen), dark or brown urine, bleeding or bruising more easily than normal (these may be signs of a liver problem)
- Spontaneous bleeding or bruising;
- Sore throat or mouth ulcers with a single episode of fever greater than 38.3°C, or above 38°C for more than one hour and/or with infection (febrile neutropenia)
- Serious infections with increased heart rate, shortness of breath or rapid breathing, fever and chills (these may be signs of a sepsis which is an infection in the blood system which may be life threatening)
- Chest pain or discomfort, changes in heart beat (fast or slow), palpitations, light headedness, fainting, dizziness, lips turning blue colour, shortness of breath, swelling (oedema) of your lower limbs or skin (these may be signs of heart problems)
- A condition called Toxic epidermal necrolysis (TEN) which starts with fever and 'flu - like symptoms. This leads to blistering peeling skin with painful raw areas
- A condition called "interstitial lung disease or pneumonitis. Symptoms include shortness of breath, cough or anxiety, confusion and restlessness
- Severe skin reactions including rash, blisters or lesions
- Fainting
- Irregular heartbeat.
VERY COMMON SIDE EFFECTS
- Tiredness, fatigue, pale skin
- Sore throat, runny nose, blocked nose, sneezing, feeling of pressure or pain in the cheeks or forehead with or without fever, cough, hoarseness, weak voice or voice loss (signs of a respiratory tract infection)
- Painful and frequent urination (signs of a urinary tract infection)
- Reduced appetite
- Shortness of breath, difficulty breathing
- Back pain
- Nausea
- Diarrhoea
- Vomiting
- Constipation
- Mouth sores or ulcers with gum inflammation
- Abdominal (belly) pain
- Hair loss or hair thinning
- Rash
- Itching
- Tiredness (fatigue)
- Weakness
- Fever
- Headache
- Swollen hands, ankles or feet
- Dizziness or light headedness
- Cough
Very common side effects that may show up in your test results
- Low level of:
- Types of white blood cells
- Red blood cells
- Platelets
- Haemoglobin
- Calcium in the blood, which may sometimes lead to cramps
- Phosphate in the blood
- Sugar in the blood
- Albumin in the blood. - High level of:
- Some liver function blood test result(s)
- Creatinine.
COMMON SIDE EFFECTS
- Abdominal pain, nausea, vomiting, diarrhoea, swelling or bloating of the abdomen and feeling sick (signs of gastroenteritis, which is an inflammation of the stomach lining)
- Spontaneous bleeding or bruising
- Watering or tearing of eyes
- Dry eye
- Strange taste in the mouth
- Upset stomach, indigestion, heartburn
- Skin reddening
- Sensation of losing balance (vertigo)
- Dry skin
- Loss of skin colour in patches (vitiligo)
- Dry mouth
- Sore throat (oropharyngeal pain).
Common side effects that may show up in your test results
- Low level of potassium in the blood, which could lead to disturbances in heart rhythm
- Change in the electrical activity of the heart.
Tell your doctor if you notice anything else that is making you unwell. Other side effects not listed above may happen in some people.
After taking KISQALI
Storage
- Keep this medicine where children cannot reach it.
A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines. - Keep your tablets in the blister until it is time to take them.
- Store the tablets in a cool dry place below 30°C.
- Do not store KISQALI in the bathroom or any other place that is hot or steamy.
- Do not leave the tablets in the car or on window sills.
Heat and dampness can destroy some medicines. KISQALI will keep well if it is cool and dry.
Disposal
If your doctor tells you to stop taking KISQALI or the tablets have passed their use by (expiry date), ask your pharmacist what to do with any that are left over.
Medicines should not be disposed of via wastewater or household waste.
Product description
What it looks like
KISQALI 200 mg
KISQALI film coated tablets are light greyish violet, unscored, round, curved with bevelled edges, debossed with "RIC" on one side and "NVR" on the other side.
The tablets are supplied in packs containing either 63, 42, or 21 tablets. All packs contain three blister strips in each carton.
63 tablets
This pack is intended for patients taking the ribociclib daily dose of 600 mg (as three tablets) once daily for 3 weeks.
Each blister strip contains 21 tablets.
42 tablets
This pack is intended for patients taking a reduced ribociclib daily dose of 400 mg (as two tablets) once daily for 3 weeks.
Each blister strip contains 14 tablets.
21 tablets
This pack is intended for patients taking the lowest ribociclib daily dose of 200 mg (one tablet once daily).
Each blister strip contains 7 tablets.
Please visit www.sparkkonnect.com.au for the Kisqali Patient Support Program.
Ingredients
Each KISQALI film coated tablet contains 200 mg of ribociclib, as the active ingredient, in the form of the succinate salt.
Each tablet also contains:
- Magnesium stearate (vegetable source) (E572)
- Microcrystalline cellulose (E460(i))
- Hyprolose (E463)
- Crospovidone (E1202)
- Colloidal silicon dioxide
- Polyvinyl alcohol (partially hydrolysed) (E1203)
- Titanium dioxide (E171)
- Iron oxide black CI77499 (E172)
- Iron oxide red CI77491 (E172)
- Iron oxide yellow CI77492 (E172)
- Purified talc (E553b)
- Lecithin (soy) (E322)
- Xanthan gum (E415).
KISQALI does not contain sucrose, lactose, gluten, tartrazine, azo dyes, or any animal products.
Sponsor
KISQALI is supplied in Australia by:
NOVARTIS Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1800 671 203
www.novartis.com.au
® = Registered trademark,
© Copyright 2019.
Australian Registration Number
AUST R 280246.
Date of preparation
This leaflet was prepared in May 2020.
Internal document code
(kis200220c_v2 is based on PI kis200220i).
Published by MIMS June 2020
 Tables 2 to 6 contain Kisqali dose modification guidelines for specific adverse reactions (also see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects)).
Tables 2 to 6 contain Kisqali dose modification guidelines for specific adverse reactions (also see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects)). In case of QTcF prolongation at any given time during treatment:
In case of QTcF prolongation at any given time during treatment:



 Clinically relevant adverse reactions reported in less than 10% of patients who received Kisqali plus AI included rash (9%), dizziness (9%), vomiting (8%), peripheral oedema (7%), pruritus (7%), dyspnoea (7%), stomatitis (6%), oropharyngeal pain (6%), hypocalcaemia (5%), hypokalaemia (4.8%), and decreased appetite (4.8%).
Clinically relevant adverse reactions reported in less than 10% of patients who received Kisqali plus AI included rash (9%), dizziness (9%), vomiting (8%), peripheral oedema (7%), pruritus (7%), dyspnoea (7%), stomatitis (6%), oropharyngeal pain (6%), hypocalcaemia (5%), hypokalaemia (4.8%), and decreased appetite (4.8%).
















 Additionally, time to progression on next-line therapy or death (PFS2) in patients in the Kisqali arm was longer compared to patients in the placebo arm (HR: 0.692 (95% CI: 0.548, 0.875)) in the overall study population. The median PFS2 was 32.3 months (95% CI: 27.6, 38.3) in the placebo arm and was not reached (95% CI: 39.4, NE) in the Kisqali arm. Similar results were observed in the NSAI sub-group (HR: 0.660 (95% CI: 0.503, 0.868); median PFS2: 32.3 months (95% CI: 26.9, 38.3) in the placebo arm vs not reached (95% CI: 39.4, NE) in the ribociclib arm).
Additionally, time to progression on next-line therapy or death (PFS2) in patients in the Kisqali arm was longer compared to patients in the placebo arm (HR: 0.692 (95% CI: 0.548, 0.875)) in the overall study population. The median PFS2 was 32.3 months (95% CI: 27.6, 38.3) in the placebo arm and was not reached (95% CI: 39.4, NE) in the Kisqali arm. Similar results were observed in the NSAI sub-group (HR: 0.660 (95% CI: 0.503, 0.868); median PFS2: 32.3 months (95% CI: 26.9, 38.3) in the placebo arm vs not reached (95% CI: 39.4, NE) in the ribociclib arm).
 The clinical benefit rate in the Kisqali plus fulvestrant arm and in the placebo plus fulvestrant arm is summarized in Table 22.
The clinical benefit rate in the Kisqali plus fulvestrant arm and in the placebo plus fulvestrant arm is summarized in Table 22.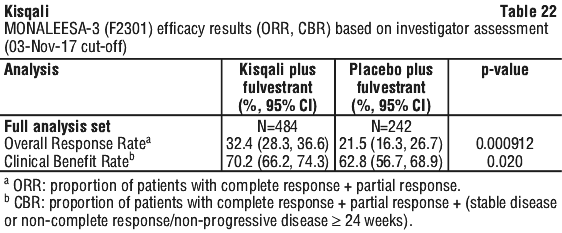

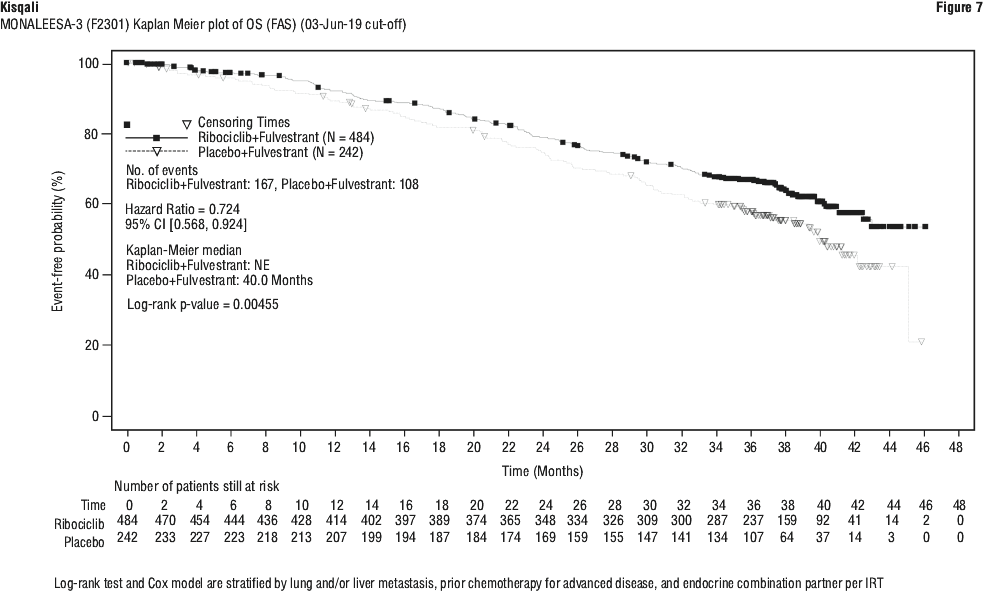 Additionally, time to progression on next-line therapy or death (PFS2) in patients in the Kisqali arm was longer compared to patients in the placebo arm (HR: 0.670 (95% CI: 0.542, 0.830)) in the overall study population. The median PFS2 was 39.8 months (95% CI: 32.5, NE) for the Kisqali arm and 29.4 months (95% CI: 24.1, 33.1) in the placebo arm.
Additionally, time to progression on next-line therapy or death (PFS2) in patients in the Kisqali arm was longer compared to patients in the placebo arm (HR: 0.670 (95% CI: 0.542, 0.830)) in the overall study population. The median PFS2 was 39.8 months (95% CI: 32.5, NE) for the Kisqali arm and 29.4 months (95% CI: 24.1, 33.1) in the placebo arm.