1 Name of Medicine
Donanemab.
2 Qualitative and Quantitative Composition
Each vial contains 350 mg/20 mL (17.5 mg/mL) donanemab.
Donanemab is a recombinant monoclonal humanised antibody produced in Chinese hamster ovary (CHO) cells and has a molecular weight of 145 kDa.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrated solution for intravenous infusion.
Sterile, non-pyrogenic, preservative free solution in a single dose vial. The solution is clear to opalescent, colourless to slightly yellow to slightly brown solution, free of visible particles.
4.1 Therapeutic Indications
Kisunla is indicated for the treatment of patients with Mild Cognitive Impairment (MCI) due to Alzheimer's disease and mild Alzheimer's dementia (early symptomatic Alzheimer's disease) that are apolipoprotein Eε4 (ApoE ε4) heterozygotes or non-carriers.
Beta amyloid evidence consistent with Alzheimer's disease (AD) should be confirmed using a validated test prior to initiating treatment.
4.2 Dose and Method of Administration
Treatment should be initiated by a physician experienced in the diagnosis and treatment of Alzheimer's disease. Kisunla should be administered in specialised centres under the supervision of a multidisciplinary team trained in detection, monitoring and management of ARIA and experienced in detecting and managing infusion related reactions.
Kisunla is single use only. Use in one patient on one occasion only. It contains no antimicrobial preservative.
Dosing information.
The recommended dose of donanemab is 350 mg for the first dose, 700 mg for the second dose, 1050 mg for the third dose (350/700/1050 mg), followed by 1400 mg every 4 weeks (see Table 1). Treatment should be maintained until amyloid plaques are cleared, as confirmed using a validated method, up to a maximum of 18 months. Treatment should be continued for up to 18 months if monitoring of amyloid plaque clearance with a validated method is not possible.
The benefit-risk of treatment should be reassessed at regular intervals on an individual basis and if the patient progresses to moderate Alzheimer's disease.
Kisunla must be diluted and is administered as an intravenous infusion over approximately 30 minutes every four weeks.

Monitoring and dosing interruption for amyloid related imaging abnormalities.
A recent (within 6 months) baseline brain magnetic resonance imaging (MRI) should be available prior to initiating treatment. An MRI should be performed prior to the second dose (one month), prior to the third dose (two months), prior to the fourth dose (usually three months) and prior to the seventh dose (usually six months) (see Section 4.4 Special Warnings and Precautions for Use).
The recommendations for dosing interruptions for patients with amyloid-related imaging abnormalities-oedema/effusions (ARIA-E) and amyloid-related imaging abnormalities haemorrhage/hemosiderin deposition (ARIA-H) are provided in Table 2.
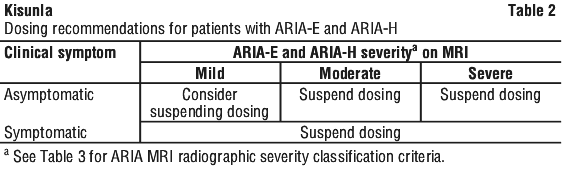 In case of asymptomatic mild ARIA, consider dose suspension based on radiological features of ARIA, number of ARIA episodes and clinical condition. In case of asymptomatic moderate or severe ARIA and symptomatic ARIA, suspend dose until MRI demonstrates radiographic resolution (ARIA-E) or stabilisation (ARIA-H) and symptoms, if present, resolve. Consider a follow-up MRI to assess for resolution (ARIA-E) or stabilisation (ARIA-H) 2 to 4 months after initial identification. Resumption of dosing or permanent discontinuation after ARIA-E resolution and ARIA-H stabilisation should be guided by clinical judgment including re-evaluation of risk factors (see Section 4.4 Special Warnings and Precautions for Use). Standard supportive treatment, including corticosteroids may be considered in case of ARIA-E.
In case of asymptomatic mild ARIA, consider dose suspension based on radiological features of ARIA, number of ARIA episodes and clinical condition. In case of asymptomatic moderate or severe ARIA and symptomatic ARIA, suspend dose until MRI demonstrates radiographic resolution (ARIA-E) or stabilisation (ARIA-H) and symptoms, if present, resolve. Consider a follow-up MRI to assess for resolution (ARIA-E) or stabilisation (ARIA-H) 2 to 4 months after initial identification. Resumption of dosing or permanent discontinuation after ARIA-E resolution and ARIA-H stabilisation should be guided by clinical judgment including re-evaluation of risk factors (see Section 4.4 Special Warnings and Precautions for Use). Standard supportive treatment, including corticosteroids may be considered in case of ARIA-E.
Kisunla should be permanently discontinued after serious ARIA-E, serious ARIA-H or intracerebral haemorrhage greater than 1 cm (see Section 4.3 Contraindications).
Radiographic severity.
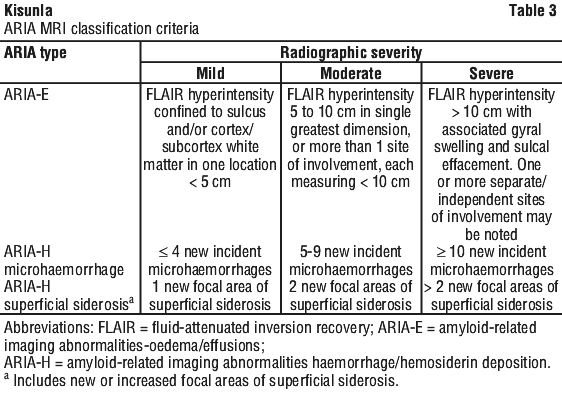
The radiographic severity of ARIA associated with donanemab was classified by the criteria shown in Table 3.
Dilution instruction.
Prior to administration, Kisunla must be diluted with 0.9% sodium chloride injection.
Use aseptic technique when preparing the Kisunla diluted solution for intravenous infusion.
Allow Kisunla to equilibrate to room temperature for approximately 30 minutes before preparation.
Parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration, whenever solution and container permit. Kisunla solution is clear to opalescent, colourless to slightly yellow to slightly brown and free of visible particles. If particulate matter or discolourations are identified, discard the vial.
Withdraw required volume of Kisunla using an appropriately sized needle and transfer to the infusion bag.
The concentrate should be diluted only in infusion bags containing sodium chloride 9 mg/mL (0.9%).
The final concentration after dilution is approximately 4 mg/mL to 10 mg/mL (see Table 4).
 Gently invert the Kisunla diluted solution to mix completely. Do not shake or freeze dosing solution.
Gently invert the Kisunla diluted solution to mix completely. Do not shake or freeze dosing solution.
Each vial is for one time use only. Use in one patient on one occasion only. Contains no antimicrobial preservative. Discard any unused portion left in the vial.
After dilution, immediate use is recommended. If the Kisunla diluted solution is not administered immediately, store refrigerated at 2°C to 8°C for up to 72 hours or at room temperature (20°C to 25°C) for up to 12 hours.
Storage times include the duration of infusion.
Administration.
Kisunla solution for infusion should be prepared and administered by a qualified healthcare professional using aseptic technique to ensure the sterility of the prepared solution:
Visually inspect the Kisunla diluted solution for particles or discolouration prior to administration. Do not use if it is discoloured, or opaque or foreign particles are seen.
Prior to infusion, if the diluted solution has been stored under refrigeration, allow the Kisunla diluted solution to warm to room temperature.
The intravenous administration set (infusion line) should be connected to the prepared intravenous bag and the line should be primed.
Administer the entire diluted solution intravenously over a period of at least 30 minutes.
Promptly discontinue the infusion upon the first observation of any signs or symptoms consistent with a hypersensitivity-type reaction (see Section 4.4 Special Warnings and Precautions for Use).
At the end of the infusion, to ensure a full dose is administered, the infusion line should be flushed with sodium chloride 9 mg/mL (0.9%) solution for injection. The flush should be administered at the same rate as used for Kisunla administration. The time required to flush Kisunla solution from the infusion line is in addition to the minimum 30 minutes infusion time.
Observe the patient post-infusion for a minimum of 30 minutes.
Missed dose.
If an infusion is missed, the missed dose should be administered at the next possible occasion. Then, resume the recommended dosing regimen every 4 weeks at the same dose as soon as possible.
Paediatric population.
There is no relevant use of Kisunla in the paediatric population for the treatment of Alzheimer's disease.4.3 Contraindications
Hypersensitivity to donanemab or to any of the excipients listed, see Section 6.1 List of Excipients.
Baseline MRI findings of prior intracerebral haemorrhage greater than 1 cm, more than 2 microhaemorrhages, superficial siderosis or vasogenic oedema (ARIA-E), which are suggestive of cerebral amyloid angiopathy (CAA).
Severe white matter disease.
Any finding that could prevent a satisfactory MRI evaluation for safety monitoring.
4.4 Special Warnings and Precautions for Use
Controlled access programme.
In order to promote the safe and effective use of Kisunla, initiation of treatment in all patients should be through a central registration system implemented as part of a controlled access programme.
Pre-treatment screening.
The efficacy and relative safety of Kisunla have been demonstrated in a population of patients with evidence of early symptomatic Alzheimer's disease based on a clinical history of cognitive decline over six months and on radiologically imaged pathological amyloid deposits in the brain (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Monoclonal antibodies directed against aggregated forms of beta amyloid, including Kisunla, can cause amyloid related imaging abnormalities (ARIA). ARIA includes amyloid related imaging abnormalities-oedema/effusions (ARIA-E; also known as cerebral vasogenic oedema) and amyloid-related imaging abnormalities haemorrhage/hemosiderin deposition (ARIA-H; includes cerebral microhaemorrhage and cortical superficial siderosis). Intracerebral haemorrhage greater than 1 cm has been observed. ARIA can occur spontaneously in patients with Alzheimer's disease, particularly in patients with MRI findings suggestive of cerebral amyloid angiopathy, such as pretreatment microhaemorrhage or superficial siderosis. ARIA-H associated with monoclonal antibodies directed against aggregated forms of beta amyloid generally occurs in association with an occurrence of ARIA-E. ARIA-H of any cause and ARIA-E can occur together.
The safety of donanemab has not been examined in patients with pre-treatment MRI showing ARIA-E, more than 4 microhaemorrhages, more than 1 area of superficial siderosis, severe white matter disease or intracerebral haemorrhage greater than 1 cm (see Section 4.3 Contraindications).
Apolipoprotein E (ApoE) genotype.
Kisunla is not indicated in ApoE ε4 homozygous patients (see Section 4.1 Therapeutic Indications). In study TRAILBLAZER-ALZ-2, 17% (143/850) of patients with known genotype in the Kisunla arm were ApoE ε4 homozygotes, 53% (452/850) were heterozygotes, and 30% (255/850) were non-carriers. The incidence of ARIA was higher in ApoE ε4 homozygotes (55% on Kisunla vs. 22% on placebo) than in heterozygotes (36% on Kisunla vs. 13% on placebo) and non-carriers (25% on Kisunla vs. 12% on placebo). Among patients treated with Kisunla, symptomatic ARIA-E occurred in 8% of ApoE ε4 homozygotes compared with 7% of heterozygotes and 4% of non-carriers. Serious events of ARIA occurred in 3% of ApoE ε4 homozygotes, 2% of heterozygotes and 1% of noncarriers. The recommendations for management of ARIA do not differ between ApoE ε4 heterozygotes and noncarriers (see Section 4.2 Dose and Method of Administration). Screening for ApoE ε4 alleles prior to treatment is required.
Cerebral amyloid angiopathy.
Neuroimaging findings that may indicate cerebral amyloid angiopathy (CAA) include evidence of prior intracerebral haemorrhage, cerebral microhaemorrhage, and cortical superficial siderosis. CAA has an increased risk for intracerebral haemorrhage. The presence of an ApoE ε4 allele is also associated with CAA.
Amyloid-related imaging abnormalities (ARIA).
Serious cases of amyloid-related imaging abnormalities (ARIA) have been observed in donanemab clinical studies and some have been fatal (see Section 4.8 Adverse Effects (Undesirable Effects)).
Most ARIA events were first observed within 24 weeks of initiation of treatment. Most serious ARIA events occurred within 12 weeks of initiation of treatment. Access to MRI should be available during the treatment period of donanemab. An MRI should be performed at baseline (within 6 months of initiating treatment), prior to the second dose, prior to third dose, prior to fourth dose and prior to the seventh dose (see Section 4.2 Dose and Method of Administration). MRI may also be indicated if ARIA symptoms occur. ARIA is often asymptomatic, although serious and life-threatening events including seizure and status epilepticus may occur. When present, symptoms may include, but are not limited to, headache, confusion, nausea, vomiting, unsteadiness, dizziness, tremor, visual disturbances, speech disturbances, worsening cognitive function, and alteration of consciousness.
Recommendations for dosing interruptions in patients with ARIA.
If symptoms of ARIA-H occur, it is often in the presence of ARIA-E and managed as for ARIA-E. The recommendations for dosing interruptions for patients with ARIA-E and ARIA-H are provided in Table 2 (see Section 4.2 Dose and Method of Administration).
Donanemab should be permanently discontinued if serious ARIA-E, serious ARIA-H or intracerebral haemorrhage greater than 1 cm occurs (see Section 4.3 Contraindications).
Concomitant antithrombotic medication.
Patients who received donanemab and an antithrombotic medicine (acetylsalicylic acid, other antiplatelets, or anticoagulants), did not have an increased frequency of ARIA. The majority of exposures to antithrombotic medicines were to acetylsalicylic acid (81%). The incidence of ARIA-H was 30% (106/349) in patients taking Kisunla with a concomitant antithrombotic medication within 30 days compared to 29% (148/504) who did not receive an antithrombotic within 30 days of an ARIA-H event.
The incidence of intracerebral haemorrhage greater than 1 cm in diameter was 0.6% (2/349 patients) in patients taking Kisunla with a concomitant antithrombotic medication compared to 0.4% (2/504) in those who did not receive an antithrombotic. The number of events and the limited exposure to non-acetylsalicylic acid antithrombotic medicines limit definitive conclusions about the risk of ARIA or intracerebral haemorrhage in patients taking antithrombotic medicines.
Because ARIA-H and intracerebral haemorrhages greater than 1 cm in diameter have been observed in patients taking donanemab, additional caution should be exercised when considering the administration of antithrombotics or a thrombolytic agent (e.g. tissue plasminogen activator) to a patient already being treated with donanemab.
In the long-term extension of the Phase 3 study, a fatal intracerebral haemorrhage occurred in a patient taking Kisunla in the setting of focal neurologic symptoms of ARIA and the use of a thrombolytic agent. Additional caution should be exercised when considering the administration of antithrombotics or a thrombolytic agent (e.g. tissue plasminogen activator) to a patient already being treated with Kisunla. Because ARIA can cause focal neurologic deficits similar to those observed in an ischaemic stroke, treating clinicians should consider whether such symptoms could be due to ARIA before giving thrombolytic therapy in a patient being treated with donanemab.
Immunogenicity.
In clinical studies, 88.1% of donanemab-treated patients developed anti-drug antibodies (ADA) and all of the patients with ADA had neutralising antibodies. Although donanemab exposure decreased with increasing ADA titre, the development of ADA was not associated with loss of clinical efficacy of donanemab. All patients reporting infusion-related reactions had ADA. Higher ADA titre was associated with increased incidence of infusion-related reactions/immediate hypersensitivity events.
Infusion related reactions.
Infusion-related reactions, including anaphylaxis have been observed with administration of Kisunla (see Section 4.8 Adverse Effects (Undesirable Effects)). These reactions may be severe or life-threatening and typically occur during infusion or within 30 minutes post infusion. Signs and symptoms of infusion-related reactions may include erythema, chills, nausea, vomiting, sweating, headache, chest tightness, dyspnoea, and changes in blood pressure. If serious infusion-related reactions occur, discontinue administration of Kisunla immediately and initiate appropriate treatment.
Sodium.
This medicinal product contains 46 mg sodium per 1400 mg dose, equivalent to 2% of the WHO recommended maximum daily intake of 2 g sodium for an adult.
Use in hepatic impairment.
Hepatic impairment did not affect the PK of Kisunla based on population PK analysis. No dose adjustment is necessary in patients with hepatic impairment.
Use in renal impairment.
Renal impairment did not affect the PK of Kisunla based on population PK analysis. No dose adjustment is necessary in patients with renal impairment.
Use in the elderly.
Clinical studies with Kisunla did not include sufficient numbers of younger adult patients to determine if patients 65 years of age and older responded differently than younger adult patients.
Paediatric use.
Safety and effectiveness of Kisunla in paediatric patients have not been established.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No drug interaction studies have been performed. No pharmacokinetic drug interactions are expected based on the characteristics of donanemab.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data on the effect of donanemab on human fertility. No animal studies have been performed to test donanemab for potential fertility impairment.
(Category B2)
There are no or limited data from the use of donanemab in pregnant women. Animal embryofetal development toxicity studies have not been conducted with donanemab. Donanemab is an IgG1-based antibody and may be transferred to the fetus during the third trimester.
Kisunla is not recommended during pregnancy.
Lactation studies have not been conducted in animals. Human immunoglobulin G (IgG) is known to be excreted in human milk; therefore, donanemab may be transmitted from the mother to the breastfed infant. The risks to a breastfed infant are unknown.4.7 Effects on Ability to Drive and Use Machines
There have been no studies conducted to determine the effects of Kisunla on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
In two placebo-controlled studies in patients with AD, a total of 984 adults received at least one dose of donanemab. Of these, 816 participants were in the indicated population. Study participants were administered three doses of 700 mg donanemab at four weekly intervals, followed by 1400 mg of donanemab at four weekly intervals for a maximum of 18 months (standard titration).
Based on ApoE ε4 carrier status, of the patients treated with donanemab, 30% (291/984) were non-carriers, 53% (522/984) were heterozygotes and 17% (168/984) were homozygotes. With the exception of events of ARIA, the safety profile was the same across genotypes.
The most frequently reported adverse reactions were ARIA-E (24.4%), ARIA-H (31.3%) and headache (13.1%). The most important serious adverse reactions were: serious ARIA-E (1.5%), serious ARIA-H (0.4%), and serious hypersensitivity including infusion-related reactions (0.6%). Anaphylaxis was uncommonly reported (0.3%). All patients reporting infusion-related reactions had ADA. Increased incidence of infusion-related reactions/immediate hypersensitivity events were associated with high ADA titre (see Section 4.4 Special Warnings and Precautions for Use). See Table 5.
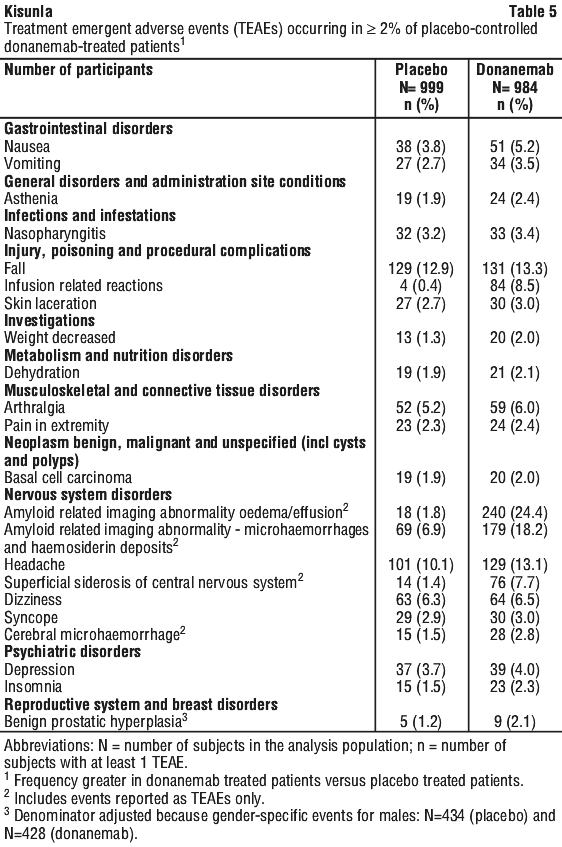
Less common adverse reactions.
Injury, poisoning and procedural complications - anaphylactic reaction (uncommon).
Description of selected adverse reactions.
Amyloid-related imaging abnormalities in the indicated population (standard titration).
ARIA (ARIA-E or ARIA-H) was observed in 32.6% (266/816) of patients treated with donanemab, compared to 12.7% (105/825) of patients on placebo in the placebo-controlled studies. Symptomatic ARIA occurred in 5.8% (47/816) of patients on donanemab. Serious ARIA events were reported for 1.3% (11/816) of patients treated with donanemab. Clinical symptoms associated with ARIA-E resolved in approximately 75% (33/44) of patients.
ARIA-E was observed in 20.8% (170/816) of patients treated with donanemab compared with 1.6% (13/825) of patients on placebo. The maximum radiographic severity for ARIA-E was mild in 6.5% of patients, moderate in 12.3% of patients, and severe in 1.7% of patients. Symptomatic ARIA-E was reported for 5.4% of patients treated with donanemab in placebo controlled clinical trials. The median time to resolution of ARIA-E was approximately 9 weeks.
ARIA-H can occur spontaneously in patients with AD independent of treatment. ARIA-H was observed in 26.7% (218/816) of patients treated with donanemab compared with 11.6% (96/825) of patients on placebo. The maximum radiographic severity for ARIA-H was mild in 14.1% of patients, moderate in 5.0% of patients, and severe in 7.5% of patients. Symptomatic ARIA-H was reported for 1.0% (8/816) of patients treated with donanemab compared with 0.2% (2/825) of patients on placebo. Isolated ARIA-H (i.e. ARIA-H in patients who did not also experience ARIA-E) was observed in 11.8% (96/816) of donanemab treated patients compared to 11.0% (91/825) on placebo.
The majority of first ARIA radiographic events in the placebo-controlled studies occurred early in treatment (within 24 weeks of initiation of treatment), although ARIA can occur at any time and patients can have more than one episode.
Phase 3 study TRAILBLAZER-ALZ-6.
The donanemab dosing regimen of 350/700/1050 mg, followed by 1400 mg every 4 weeks was evaluated in a phase 3b multicentre, randomised, double-blind, study in adults with early symptomatic AD (MCI due to AD or mild AD dementia, MMSE score 20 to 28 inclusive) and evidence of amyloid beta pathology confirmed by amyloid PET scan.
843 patients were randomised at a 1:1:1:1 ratio into four donanemab dosing regimens for a total of 72 weeks, 700 mg for the first three infusions, then 1400 mg every 4 weeks thereafter (n=207; standard titration), or one of the three alternative dosing regimens (including the dosing regimen: 350/700/1050 mg, followed by 1400 mg every 4 weeks; n=212; modified titration).
The primary endpoint of the study was the proportion of participants with any occurrence of ARIA-E by week 24. The results showed that 14% of patients receiving 350/700/1050 mg (modified titration), compared with 24% receiving 700/700/700 mg (standard titration), experienced any occurrence of ARIA-E by week 24, a 41% relative risk (Table 6) reduction. Similar amyloid plaque reductions were seen at 24 weeks in all dosing regimens.
 The secondary endpoint of the study was the proportion of participants with any occurrence of ARIA-H by week 24. The results showed that 20% of patients receiving modified titration, compared with 25% receiving standard titration, experienced any occurrence of ARIA-H by week 24, a 18% relative risk reduction. Symptomatic ARIA-E was reported for 2.8% of patients treated with donanemab with modified titration during the 76-week treatment period. See Table 7.
The secondary endpoint of the study was the proportion of participants with any occurrence of ARIA-H by week 24. The results showed that 20% of patients receiving modified titration, compared with 25% receiving standard titration, experienced any occurrence of ARIA-H by week 24, a 18% relative risk reduction. Symptomatic ARIA-E was reported for 2.8% of patients treated with donanemab with modified titration during the 76-week treatment period. See Table 7.

ApoE ε4 carrier status and risk of ARIA.
In placebo-controlled studies, the incidence of ARIA was lower in non-carriers (24.1% donanemab vs 11.3% placebo) and heterozygotes (37.4% donanemab vs. 13.4% placebo) than in homozygotes (58.3% donanemab vs 21.3% placebo). Among patients treated with donanemab, symptomatic ARIA-E occurred in 4.1% of non-carriers and 6.1% of heterozygotes compared with 7.7% of homozygotes. Serious events of ARIA occurred in approximately 0.7% of non-carriers, 1.7% heterozygotes and 3% of homozygotes. Among patients treated with donanemab, the rate of severe radiographic ARIA-E was lower in noncarriers 1.0% (3/291) and heterozygotes 2.1% (11/522) compared to homozygotes 4.2% (7/168). The rate of severe radiographic ARIA-H was lower in non-carriers 4.5% (13/291) and heterozygotes 9.2% (48/522) compared to homozygotes 24.4% (41/168).
Among the patients who experienced an event of ARIA-E and continued on donanemab with or without dose interruption, the rates of recurrence were 32.4% (11/34) in non-carriers, 26.7% (27/101) in heterozygotes and 28.6% (14/49) in homozygotes.
Among the patients who experienced an event of ARIA-H and continued on donanemab with or without dose interruption, the rates of recurrence were 35.1% (13/37) in non-carriers (compared with 31.8% [7/22] on placebo), 39.1% (45/115) in heterozygotes (compared with 38.6% [17/44] on placebo), and 51.7% (31/60) in homozygotes (compared with 30.8% [8/26] on placebo).
Intracerebral haemorrhage.
In placebo-controlled studies, intracerebral haemorrhage greater than 1 cm has been observed after treatment with donanemab in 0.3% compared to 0.2% for placebo. Additionally, in the pivotal study, a concurrent intracerebral haemorrhage with fatal ARIA-H has been observed in a patient treated with donanemab (see Section 4.4 Special Warnings and Precautions for Use).
In Study TRAILBLAZER-ALZ 6, intracerebral haemorrhage greater than 1 cm was reported in 1% (2/212) of patients treated with donanemab for 24 weeks at the dosing regimen of 350/700/1050 mg, followed by 1400 mg every 4 weeks.
Infusion-related reactions.
Infusion reactions were observed in 8.5% of patients treated with donanemab compared to 0.4% on placebo. Anaphylaxis was uncommonly reported (0.3%). Serious infusion reactions or hypersensitivity occurred in 0.6% of patients treated with donanemab compared to 0.2% on placebo.
The majority of infusion reactions and hypersensitivity reactions have occurred within the first 4 doses of donanemab, although they can occur at any time.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Single doses up to 40 mg/kg (approximately 2800 mg in a 70 kg person) have been administered.
ARIA-E occurred in two out of four patients administered this dose and resolved. In case of an overdose, initiate supportive therapy if necessary.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Reductions in cerebral amyloid plaques, as measured by amyloid positron emission tomography (PET), were observed among patients receiving donanemab. Donanemab reduced tau pathophysiology, as measured by plasma P-tau217.
Mechanism of action.
Donanemab is an immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against an insoluble, pyroglutamate N terminal truncated form of amyloid beta (N3pE Aβ) present only in brain amyloid plaques. Donanemab binds to N3pE Aβ and aids plaque removal through microglial-mediated phagocytosis.
Biomarkers.
The percentage of donanemab treated patients with amyloid clearance (that is, less than 24.1 Centiloids or visually negative on an amyloid PET scan) in Study TRAILBLAZER-ALZ 2 is represented in Figure 1.
A reduction in plasma P-tau217 (log 10) was observed with donanemab compared to placebo. In a study population with low to medium levels of brain tau (baseline SUVr ≤ 1.46), LS mean change difference ± SE was -0.19 ± 0.011 and -0.25 ± 0.014 at Weeks 24 and 76, respectively, compared to placebo (p < 0.0001 at both time points). Consistent with this, in a study population with low to medium and high levels of brain tau, LS mean change difference ± SE was - 0.16 ± 0.010 and -0.22 ± 0.012 at Weeks 24 and 76, respectively, compared to placebo (p < 0.0001 at both time points).

Clinical trials.
Clinical efficacy and safety. The efficacy and safety of donanemab were evaluated in a Phase 3 (TRAILBLAZER-ALZ 2) and a Phase 2 (TRAILBLAZER-ALZ) study, both double-blind placebo controlled, parallel group, in patients with early symptomatic AD (Mild Cognitive Impairment (MCI) or mild dementia due to AD) and evidence of amyloid beta pathology confirmed by amyloid PET scan. The participants also had evidence of pathologic tau deposition on a flortaucipir PET scan. The Phase 3 study confirmed the efficacy and safety results observed in the Phase 2 Study. For the safety analysis, patients were followed for up to 76 weeks or end of treatment plus 57 days.
Phase 3 Study TRAILBLAZER-ALZ 2. In this study, 1736 patients were randomised 1:1 to receive 700 mg of donanemab every 4 weeks for the first 3 doses, and then 1400 mg every 4 weeks via intravenous infusion (N = 860) or placebo (N = 876) for a total of up to 72 weeks. The study enrolled men or women, aged 60-85 years with gradual and progressive change in memory function for 6 months or more, a MMSE score of 20 to 28 at screening, who met the florbetapir F18 or florbetaben F18 scan criteria and flortaucipir F18 scan criteria. Patients were included in the study based on visual assessment of tau PET imaging with flortaucipir and quantitation by standardised uptake value ratio (SUVR). The low-medium tau level population included patients with tau SUVR ratios of 1.10 to 1.46, inclusive, with a topographic deposition pattern consistent with moderate AD or ≤ 1.46 with a topographic deposition pattern consistent with advanced AD. The high tau level population included patients with tau SUVR ratio > 1.46 with a topographic deposition pattern consistent with either moderate or advanced AD. The study includes a double-blind extension period of 78 weeks duration. Dosing was continued until study completion or amyloid plaque was cleared, defined as demonstrating a plaque level of less than 25 Centiloids for two consecutive amyloid PET scans or a single PET scan demonstrating a plaque level of less than 11 Centiloids. Additionally, dose suspension was allowed for treatment-emergent ARIA. If patients were already on symptomatic treatment (acetylcholinesterase inhibitors (AChEI) and/or the N-methyl-D-aspartate inhibitor, memantine) at study entry, these treatments could continue. Symptomatic treatments could be added or changed during the study, at the investigator's discretion. The study excluded patients with pre-existing ARIA-E, greater than 4 microhaemorrhages, more than 1 area of superficial siderosis, any intracerebral haemorrhage > 1 cm or severe white matter disease. Patients with significant neurological disease affecting the central nervous system other than AD that may affect cognition, including other dementias, were excluded. Patients with no or very low tau pathology were excluded from the randomised placebo-controlled portion of the study.
At baseline, 266 participants had MCI and 1040 had mild AD. Of the total number of patients randomised, 29% (510/1736) were ApoE ε4 non-carriers, 54% (930/1736) were heterozygotes, and 17% (289/1736) were homozygotes. Mean age was 73 years, with a range of 59 to 86 years, with a mean (SD) baseline weight of 71.7 kg (15.7), with a gradual and progressive change in memory function for at least 6 months and a Mini-Mental State Examination (MMSE) score of 20 to 28 (inclusive). 57.4% were female, 91.5% were White, 5.7% were of Hispanic or Latino ethnicity, 6.0% were Asian, and 2.3% were Black. 55.6% of patients were on AChEI, and 20.3% on memantine. 61% of patients were on either AChEI or memantine use.
There were two primary analysis populations based on tau PET imaging at screening with flortaucipir: 1) low medium tau level population, and 2) combined population (low medium plus high tau level population).
The primary efficacy endpoint was change in cognition and function as measured by the integrated Alzheimer's Disease Rating Scale (iADRS) score from baseline to 76 weeks. The iADRS is an integrated assessment of cognition and daily function comprised of items from the Alzheimer's Disease Assessment Scale-Cognitive subscale (ADAS Cog13) and the Alzheimer's Disease Cooperative Study - instrumental Activities of Daily Living (ADCS-iADL) scale, measuring the core domains across the AD clinical continuum. The total score ranges from 0 to 144, with lower scores reflecting worse cognitive and functional performance. Other efficacy endpoints included Clinical Dementia Rating Scale - Sum of Boxes (CDR-SB), ADAS-Cog13, ADCS-iADL.
Treatment with donanemab statistically significantly slowed clinical decline compared to placebo at week 76, with consistency across measures of cognition and function (Figure 2 and Table 8).
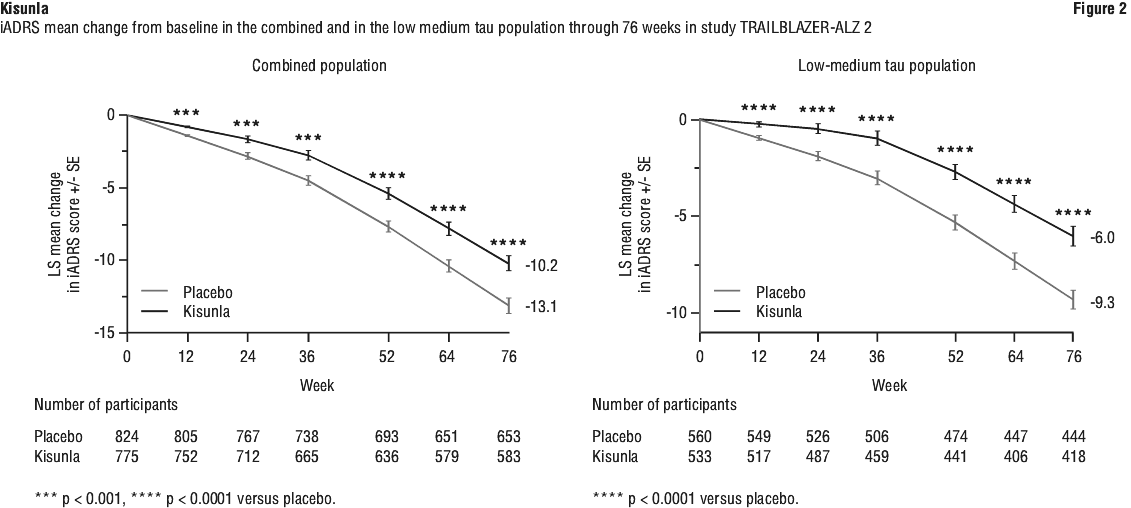
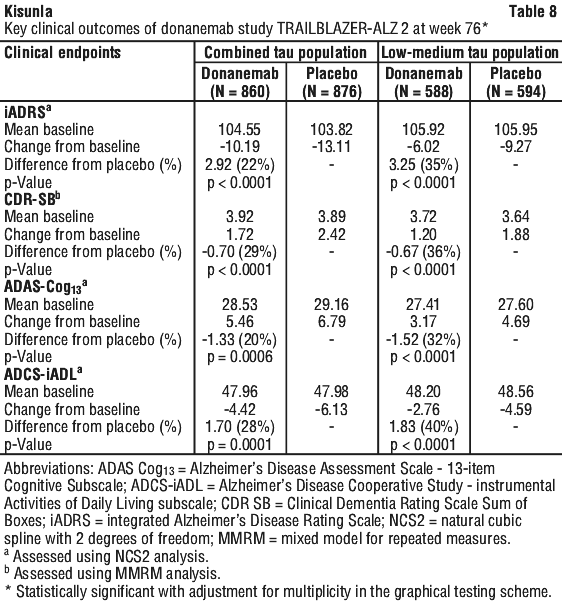 Patients treated with donanemab also had a 39% and 37% lower risk of progressing to the next stage of disease as measured by the CDR-global score (HR: 0.61, p < 0.001; and HR: 0.63, p < 0.0001) through Week 76 in the low-medium tau and in the combined population, respectively.
Patients treated with donanemab also had a 39% and 37% lower risk of progressing to the next stage of disease as measured by the CDR-global score (HR: 0.61, p < 0.001; and HR: 0.63, p < 0.0001) through Week 76 in the low-medium tau and in the combined population, respectively.
At Week 76, donanemab treatment delayed disease progression by 4.4 months and 7.5 months as assessed by iADRS and CDR-SB respectively in the low medium tau population, and by 1.4 months and 5.4 months as assessed by iADRS and CDR-SB respectively in the combined population.
At Week 76, patients treated with donanemab had less decline in cognition than placebo treated participants as assessed by the MMSE change from baseline values in both the low medium tau and in the combined population.
High tau population.
In the high-tau population (271 patients on donanemab and 281 patients on placebo), donanemab slowed clinical decline by 6% (1.26 ± 1.54 [p = 0.415]) on iADRS, and 21% (- 0.69 ± 0.25 [p = 0.006]) on CDR-SB, at Week 76 compared with placebo.
Phase 2 study TRAILBLAZER-ALZ. In this study, patients were randomised to receive 700 mg of donanemab every 4 weeks for the first 3 doses, and then 1,400 mg every 4 weeks or placebo for a total of up to 72 weeks. A total of 257 participants were randomised 1:1 to donanemab (n = 131) or placebo (n = 126). The study enrolled men or women, aged 60-85 years with gradual and progressive change in memory function for 6 months or more, a MMSE score of 20 to 28 at screening, who met the florbetapir F18 scan criteria and flortaucipir F18 scan criteria. Patients were included in the study based on visual assessment of tau PET imaging with flortaucipir and quantification by standardised uptake value ratio (SUVR). The study enrolled a low-medium tau population of patients with tau SUVR ratios of 1.10 to 1.46, inclusive, or < 1.10 if topographic deposition pattern consistent with advanced AD. In participants who were treated with donanemab, if the amyloid plaque level as assessed by florbetapir PET was 11 to less than 25 Centiloids, the dose was lowered to 700 mg. If the amyloid plaque level was less than 11 Centiloids on any one scan or was 11 to less than 25 Centiloids on two consecutive scans, donanemab was switched to placebo. Additionally, dose suspension was allowed for treatment-emergent ARIA. If patients were already on symptomatic treatment (acetylcholinesterase inhibitors (AChEI) and/or the N-methyl-D-aspartate inhibitor, memantine) at study entry, these treatments could continue. Symptomatic treatments could be added or changed during the study, at the investigator's discretion. The study excluded patients with pre-existing ARIA-E, greater than 4 microhaemorrhages, more than 1 area of superficial siderosis, any intracerebral haemorrhage > 1 cm or severe white matter disease. Patients with significant neurological disease affecting the central nervous system other than AD that may affect cognition, including other dementias, were excluded. Patients with no or very low tau pathology and high tau pathology were excluded from the study.
At baseline, 46 participants had MCI and 185 had mild AD. Of the total number of participants randomised, 26% (68/257) were ApoE ε4 non-carriers, 52% (134/257) were heterozygotes, and 21% (53/257) were homozygotes. Mean age was 75.2 years, with a range of 61 to 86 years.
48% of patients were male and 95% were white. Patients treated with donanemab demonstrated reduced clinical decline, as evidenced by a statistically significant treatment effect on change from baseline in iADRS compared to placebo at week 76 (3.20 [-32%], p=0.042).
5.2 Pharmacokinetic Properties
Absorption.
Kisunla is for intravenous administration only.
Distribution.
Following intravenous dosing, Kisunla undergoes biphasic elimination. The central volume of distribution is 3.36 L with 18.7% inter-individual variability. Peripheral volume of distribution is 4.83 L, with 93.9% inter-individual variability.
Metabolism.
Donanemab is a monoclonal antibody and is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as an endogenous IgG.
Excretion.
The half-life of Kisunla is approximately 12.1 days. Kisunla clearance was 0.0255 L/h (24.9% inter-individual variability).
Special populations.
The PK of Kisunla was not affected by age, sex, or race, based on a population PK analysis. While body weight was found to influence both clearance and volume of distribution, the resulting changes do not suggest a need for dose adjustment.
5.3 Preclinical Safety Data
Genotoxicity.
No studies have been performed to test donanemab for potential of genotoxicity. As a high molecular weight protein, donanemab is not expected to interact directly with DNA or other chromosomal material.
Carcinogenicity.
No animal studies have been performed to test donanemab for potential carcinogenicity. A weight-of-evidence assessment of all data showed a low potential for risk of carcinogenicity.6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium citrate dihydrate, citric acid, sucrose, polysorbate 80, water for injections.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Unopened vial.
Refrigerate at 2°C to 8°C until time of use.
Keep the vial in the outer carton in order to protect from light.
Do not freeze or shake.
May be stored unrefrigerated for up to 3 days at room temperature (20°C to 25°C).
Diluted solution.
Use prepared dosing solution immediately.
If not used immediately, store Kisunla dosing solution under refrigeration for up to 72 hours at 2°C to 8°C or for 12 hours at room temperature (20°C to 25°C) assuming dilution has taken place using aseptic techniques.
Storage times include the duration of infusion.
Discard unused portion.
Do not freeze Kisunla dosing solution.
6.5 Nature and Contents of Container
Kisunla 350 mg/20 mL is supplied as a clear glass vial with a rubber stopper and aluminum seal. The stopper is not made with natural rubber latex.
Kisunla is available in pack size of 1 vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
1931944-80-7.7 Medicine Schedule (Poisons Standard)
Schedule 4.
Summary Table of Changes
