What is in this leaflet?
This leaflet answers some common questions about KODATEF tablets.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of you taking KODATEF tablets against the benefits expected for you.
Pharmaceutical companies cannot give you medical advice or an individual diagnosis.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What KODATEF is used for
KODATEF is used to prevent malaria in adults for up to six months.
KODATEF is recommended for individuals travelling to areas where malaria is endemic. KODATEF contains the active ingredient tafenoquine.
KODATEF works by killing the parasites that cause malaria.
KODATEF is not addictive.
Ask your doctor if you have any questions about why KODATEF has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you take KODATEF
When you must not take KODATEF
Do not take KODATEF if:
- You have been diagnosed or suspect you may have a medical condition known as Glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency).
Persons with G6PD deficiency have an increased risk for damage to their red blood cells if they are exposed to certain foods or medicines, including KODATEF. G6PD deficiency is an inherited condition that affects different percentages of individuals based on their ethnicity: about 10% of Africans, 2% of Southeast Asians, and 10% of persons of Mediterranean background (Italian, Greek, or Middle Eastern). If you are unsure whether you have G6PD deficiency, please speak to your doctor. To determine whether you have G6PD deficiency you will need a blood test.
- You have had an allergic reaction to any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
shortness of breath.
wheezing or difficulty in breathing.
swelling of the face, lips, tongue or other parts of the body.
rash, itching or hives on the skin.
- You have severe liver disease.
- The expiry date (EXP) printed on the package has passed.
If you take this medicine after the expiry date has passed, it may not work as well.
- The packaging is torn or damaged.
Do not take KODATEF if the package is torn or shows signs of tampering. If the tamper proof seal has been pealed back or damaged, do not use. Return the damaged packaging to your pharmacist.
- You are pregnant.
- Do not give this medicine to adolescents or children under the age of 18 years or elderly patients older than 65 years of age.
Safety and effectiveness in children younger than 18 years of age or elderly persons over 65 years of age has not been established.
If you are not sure if you should be taking KODATEF, talk to your doctor or pharmacist.
Before you start to take KODATEF
Tell your doctor if:
- You have or have had any health problems, especially the following:
liver problems.
kidney problems.
- Known or suspected Glucose-6-phosphate dehydrogenase deficiency (G6PD).
- You plan to take or are currently taking metformin or other medicine for type 2 diabetes.
- You plan to take or are currently taking a medicine to suppress abnormal rhythms of the heart such as procainamide
- You plan to take or are currently taking dapsone for leprosy.
- You are pregnant or plan to become pregnant.
- You are breastfeeding or intend to breastfeed.
KODATEF may pass into breast milk and may affect your baby; therefore, your doctor will discuss the risks and benefits of taking KODATEF.
- You have a psychotic disorder such as schizophrenia.
- If you have not told your doctor about any of the above, do so before starting KODATEF.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket, or health food shop.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking KODATEF.
How to take KODATEF
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
How much KODATEF to take
Take KODATEF exactly as your doctor has prescribed. Your doctor will tell you how many KODATEF tablets to take and how often to take them.
Prevention of malaria.
Before entering the malarious area take TWO tablets once daily for THREE days.
After entering the malarious area take TWO tablets of KODATEF once WEEKLY, on the same day of the week, starting ONE WEEK after the last dose taken before entering the malarious area.
After leaving the malarious area take TWO tablets within ONE WEEK of returning. This day should be the same WEEKLY day you took your tablets while in the malarious area.
If you leave the malarious area before starting the weekly dosing regimen, take one last dose 7 days after you finish the pretravel three-day dosing regimen.
Individuals need to complete the full course of KODATEF including starting and final doses.
If you are not sure what to do, ask your doctor or pharmacist.
How to take KODATEF
Swallow tablets whole with a full glass of water.
When to take KODATEF
KODATEF can be taken with or without food.
How long to take KODATEF
Continue taking KODATEF for as long as your doctor tells you to.
KODATEF should be taken as prescribed shortly before entering a malarious area, while still in a malarious area, and for ONE week upon leaving this area. If you leave the malarious area before starting the weekly dosing regimen, take one last dose 7 days after you finish the pre-travel three-day dosing regimen.
Your doctor will advise you of the areas of the world where you may be at risk of contracting malaria.
KODATEF can be taken for up to six months for the prevention of malaria.
If you stay in a malarious area for more than six months, your doctor will tell you what to do to prevent malaria infection.
If you forget to take KODATEF
If one of the weekly KODATEF doses is missed, replace with one KODATEF dose on any day up to the time of the next weekly dose. If two of the weekly KODATEF doses are missed, replace with one KODATEF dose on the day before the next weekly dose. If three or more KODATEF doses are missed, replace with two daily doses before the next weekly dose.
If you think you may have trouble remembering your dose, ask your pharmacist for some hints.
If you take too much (overdose)
If you think that you or anyone else may have taken too much KODATEF, immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26 in Australia) for advice, or go to Accident and Emergency at your nearest hospital. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you are not sure what to do, contact your doctor or pharmacist.
While you are taking KODATEF
Things you must do
Tell all doctors, dentists and pharmacists who are treating you that you are taking KODATEF.
Tell your doctor immediately if you become pregnant while taking KODATEF.
Women of child-bearing potential should use effective contraception while taking KODATEF and for at least three months after taking the last dose.
If you have a psychotic disorder and your symptoms worsen while taking KODATEF, seek medical attention. As malaria can be a fatal disease, you should not stop taking KODATEF without telling your doctor.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed.
Be sure to keep all of your appointments with your doctor so that your progress can be checked.
Things you must not do
Do not stop taking KODATEF or change the dose without first checking with your doctor.
Do not let yourself run out of KODATEF while in a malarious area.
Do not give KODATEF to anyone else, even if they are travelling with you.
Tell your doctor if you notice any of the following and they worry you.
These are the more common or general mild side effects:
- stomach pain or discomfort
- diarrhoea
- eye deposits.
These symptoms will go away after you stop taking KODATEF.
KODATEF may cause eye deposits (swirls in the cornea of the eye that can be seen during an eye examination). These do not affect vision (including night vision) and will go away after you stop taking KODATEF.
Stop taking KODATEF and tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you experience any of the following:
seizure (fit) or convulsion.
shortness of breath.
loss of consciousness.
looking pale and yellowing of the skin and/or eyes.
blue or purple coloration of the skin.
abnormal paleness or lack of color of the skin.
feeling of confusion.
fast or abnormal heart rate.
dark colored urine.
pinkish, itchy swellings on the skin, also called hives or nettle rash.
These are serious side effects. You may need urgent medical attention. Serious side effects are rare.
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list.
After taking KODATEF
Storage
Keep your tablets in the blister pack until it is time to take them. If you take the tablets out of the blister pack they will not keep well.
Keep KODATEF in a cool dry place where the temperature stays below 30°C.
Do not leave it in the car or on window sills. Heat can destroy some medicines.
Keep KODATEF where children cannot reach it. A locked cupboard at least one-and-a-half meters above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking KODATEF, or the tablets have passed their expiry date, ask your pharmacist what to do with any tablets that are left over.
Product Description
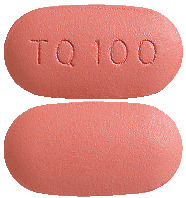
What KODATEF looks like
KODATEF tablets are dark pink, capsule shaped, debossed with “TQ100” on one side and plain on the other.
KODATEF tablets (100 mg) are supplied in blister packs. Each blister pack contains 8 tablets. There are one or two blister packs per carton (8 or 16 tablets per carton).
Ingredients
Each KODATEF tablet contains the active ingredient tafenoquine (100 mg) in the form of salt (125.6 mg tafenoquine succinate).
KODATEF tablets also contain:
Microcrystalline Cellulose
Mannitol
Magnesium Stearate
Hypromellose
Titanium dioxide
Iron oxide red and
Macrogol 400
KODATEF does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
The Australian Registration Number is AUST R: 293438
This is not all the information available on KODATEF tablets. If you have any more questions or are unsure about anything, ask your doctor or pharmacist.
Manufacturer
KODATEF is distributed in Australia by:
Biocelect Pty Ltd
Level 29,
66 Goulburn Street,
SYDNEY,
NSW 2000
Customer enquiries and Medical Information: 1300 848 628
Website: www.biocelect.com/products/kodatef
KODATEF is a registered trademark of 60 Degrees Pharmaceuticals LLC.
This leaflet was prepared on:
17 May 2022
Published by MIMS September 2022
 Individuals need to complete the full course of Kodatef including loading and terminal doses. If leaving the malarious area before the start of the maintenance regimen, a single terminal dose should be taken 7 days after the last dose of the loading regimen.
Individuals need to complete the full course of Kodatef including loading and terminal doses. If leaving the malarious area before the start of the maintenance regimen, a single terminal dose should be taken 7 days after the last dose of the loading regimen.
 Adverse reactions occurring in ≥ 1% of subjects in the Kodatef group in the placebo-controlled pooled data from Trials 043, 045, 030 and 057 are presented in Table 4.
Adverse reactions occurring in ≥ 1% of subjects in the Kodatef group in the placebo-controlled pooled data from Trials 043, 045, 030 and 057 are presented in Table 4.
 In the 24 week follow up phase after leaving the endemic region, and after receiving no further drug (tafenoquine group), or standard post-exposure prophylaxis with primaquine (mefloquine group), there were four cases of Pv malaria in the tafenoquine group and one case of Pv malaria in the mefloquine group (Table 6).
In the 24 week follow up phase after leaving the endemic region, and after receiving no further drug (tafenoquine group), or standard post-exposure prophylaxis with primaquine (mefloquine group), there were four cases of Pv malaria in the tafenoquine group and one case of Pv malaria in the mefloquine group (Table 6). The failure rate due to Pv relapse was 0.9% for the tafenoquine group and 0.7% for the primaquine group. The time to relapse after the last dose of tafenoquine or mefloquine was 12-20 weeks for the 4 tafenoquine failures and 12 weeks for the 1 mefloquine-then-primaquine failure. All 5 cases were Pv malaria.
The failure rate due to Pv relapse was 0.9% for the tafenoquine group and 0.7% for the primaquine group. The time to relapse after the last dose of tafenoquine or mefloquine was 12-20 weeks for the 4 tafenoquine failures and 12 weeks for the 1 mefloquine-then-primaquine failure. All 5 cases were Pv malaria.

