1 Name of Medicine
Selumetinib sulfate.
2 Qualitative and Quantitative Composition
Each 10 mg hard capsule contains 10 mg of selumetinib (as sulfate).
Each 25 mg hard capsule contains 25 mg of selumetinib (as sulfate).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Capsule, hard.
Koselugo 10 mg hard capsule.
White to off-white, opaque, size 4 hard capsule, banded and marked with "SEL 10" in black ink.
Koselugo 25 mg hard capsule.
Blue, opaque, size 4 hard capsule, banded and marked with "SEL 25" in black ink.4.1 Therapeutic Indications
Koselugo is indicated for the treatment of adult and paediatric patients aged 2 years and older, with neurofibromatosis type 1 (NF1) who have symptomatic, inoperable plexiform neurofibromas (PN).
4.2 Dose and Method of Administration
Therapy should be initiated by a physician experienced in the diagnosis and the treatment of patients with NF1 related tumours.
Dosage.
The recommended dosage of Koselugo is 25 mg/m2 of body surface area (BSA), taken orally twice daily (approximately every 12 hours) until disease progression or unacceptable toxicity.
Dosing in adult and paediatric patients is individualised based on BSA (mg/m2) and rounded to the nearest achievable 5 mg or 10 mg dose (up to a maximum single dose of 50 mg). Different strengths of Koselugo capsules can be combined to attain the desired dose (see Table 1).
 Treatment with Koselugo should continue as long as clinical benefit is observed, or until PN progression or the development of unacceptable toxicity.
Treatment with Koselugo should continue as long as clinical benefit is observed, or until PN progression or the development of unacceptable toxicity.
Method of administration.
Koselugo can be taken with or without food.
Koselugo capsules should be swallowed whole with water, and should not be chewed, dissolved, or opened.
Do not administer to patients who are unable or unwilling to swallow a whole capsule. Patients should be assessed for their ability to swallow a capsule before starting treatment. Standard medicine swallowing techniques are expected to be sufficient to swallow selumetinib capsules. For patients who have difficulties swallowing the capsule, referral to an appropriate healthcare professional such as a speech and language therapist could be considered to identify suitable methods that can be tailored to the particular patient.
Missed dose.
If a dose of Koselugo is missed, it should only be taken if it is more than 6 hours until the next scheduled dose.
Vomiting.
Do not take an additional dose if vomiting occurs after Koselugo administration but continue with the next scheduled dose.
Dose adjustments.
For adverse reactions. Interruption and/or dose reduction or permanent discontinuation of Koselugo may be required based on individual safety and tolerability (see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects)).
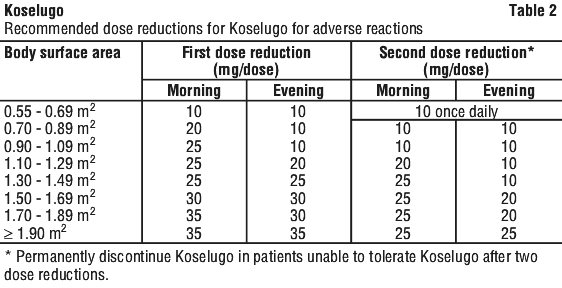
The recommended dose reductions for adverse reactions are provided in Table 2 and may require the daily dose to be divided into two administrations of different strength or for treatment to be given as a once daily dose.
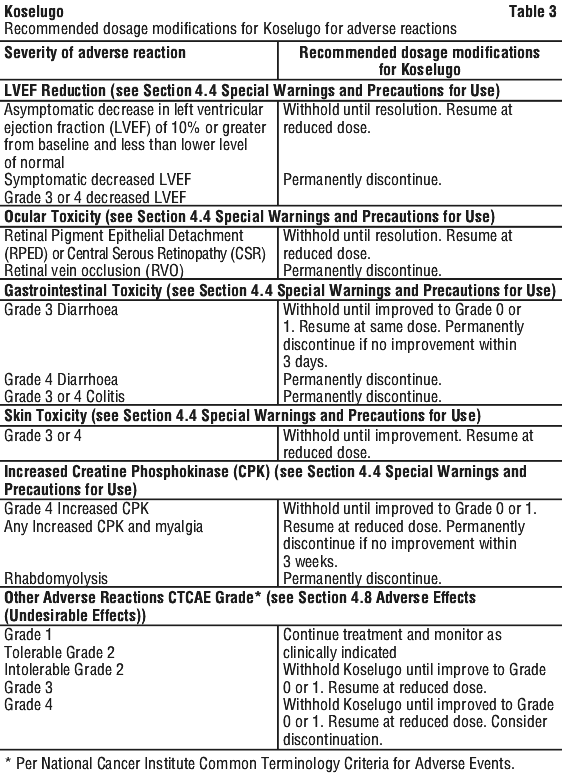
 Dosage modifications for adverse reactions are in Table 3.
Dosage modifications for adverse reactions are in Table 3.
 For drug interactions.
For drug interactions. Co-administration with CYP3A4 or CYP2C19 inhibitors.
Concomitant use of strong or moderate CYP3A4 or CYP2C19 inhibitors is not recommended and alternative agents should be considered. If a strong or moderate CYP3A4 or CYP2C19 inhibitor must be co-administered, the recommended Koselugo dose reduction is as follows:
If a patient is currently taking 25 mg/m2 twice daily, dose reduce to 20 mg/m2 twice daily.
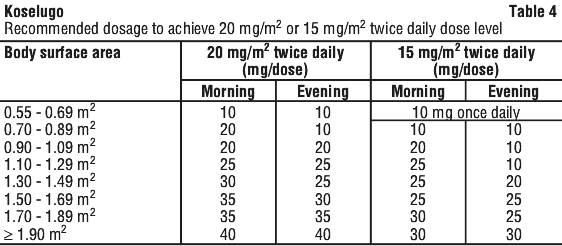
If a patient is currently taking 20 mg/m2 twice daily, dose reduce to 15 mg/m2 twice daily (see Table 4; see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Special patient populations.
Renal impairment.
Based on clinical studies no dose adjustment is recommended in patients with mild, moderate, severe renal impairment or those with End Stage Renal Disease (ESRD) (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
Based on clinical studies, no dose adjustment is recommended in patients with mild hepatic impairment. The starting dose should be reduced in patients with moderate hepatic impairment to 20 mg/m2 BSA, twice daily (see Table 4). Koselugo is not recommended for use in patients with severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Ethnicity.
Increased systemic exposure has been seen in adult Asian subjects, although there is considerable overlap with Western subjects when corrected for body weight. No specific adjustment to the starting dose is recommended for Asian patients, however, these patients should be closely monitored for adverse events (see Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients (see Section 6.1 List of Excipients).
Severe hepatic impairment.
4.4 Special Warnings and Precautions for Use
LVEF reduction.
LVEF reduction have been reported in both paediatric and adult patients (see Section 4.8 Adverse Effects (Undesirable Effects)). A small number of serious reports of LVEF reduction associated with selumetinib have been reported in paediatric patients who participated in an expanded access program.
Patients with a history of impaired left ventricular function or a baseline LVEF below institutional LLN have not been studied. LVEF should be evaluated before initiation of treatment to establish baseline values. Prior to starting Koselugo treatment, patients should have an ejection fraction above the institutional LLN.
Evaluate LVEF at approximately 3 month intervals, or more frequently as clinically indicated, during treatment. Reduction in LVEF can be managed using treatment interruption, dose reduction or treatment discontinuation (see Section 4.2 Dose and Method of Administration).
Ocular toxicity.
Advise patients to report any new visual disturbances. Adverse events of blurred vision have been reported in patients receiving Koselugo. Isolated cases of retinal pigment epithelial detachment (RPED), central serous retinopathy (CSR) and retinal vein occlusion (RVO) in adult patients with multiple tumour types, receiving treatment with Koselugo monotherapy and in combination with other anti-cancer agents, and in a single paediatric patient with pilocytic astrocytoma on Koselugo monotherapy, have been observed (see Section 4.8 Adverse Effects (Undesirable Effects)).
In line with clinical practice an ophthalmological evaluation prior to treatment initiation and at any time a patient reports new visual disturbances is recommended. In patients diagnosed with RPED or CSR without reduced visual acuity, ophthalmic assessment should be conducted every 3 weeks until resolution. If RPED or CSR is diagnosed and visual acuity is affected Koselugo therapy should be interrupted and the dose reduced when treatment is resumed (see Section 4.2 Dose and Method of Administration, Table 2). If RVO is diagnosed, treatment with Koselugo should be permanently discontinued (see Section 4.2 Dose and Method of Administration, Table 3).
Liver laboratory abnormalities.
Liver laboratory abnormalities, specifically AST and ALT elevations, can occur with Koselugo (see Section 4.8 Adverse Effects (Undesirable Effects)). Liver laboratory values should be monitored before initiation of selumetinib and at least monthly during the first 6 months of treatment, and thereafter as clinically indicated. Liver laboratory abnormalities should be managed with dose interruption, reduction or treatment discontinuation (see Section 4.2 Dose and Method of Administration, Table 2).
Gastrointestinal toxicity.
Gastrointestinal toxicities, primarily including diarrhoea, vomiting, nausea, and stomatitis occurred in patients who received Koselugo (see Section 4.8 Adverse Effects (Undesirable Effects)) and led to dose interruption, dose reduction and permanent discontinuation in some patients.
Serious gastrointestinal toxicities, including perforation, colitis, ileus, and intestinal obstruction, occurred in an unapproved population of adult patients with multiple tumour types who received Koselugo as a single agent or in combination with other anti-cancer agents. Colitis occurred in an unapproved population of paediatric patients with multiple tumour types who received Koselugo as a single agent.
Advise patients to start an anti-diarrhoeal agent (e.g. loperamide) immediately after the first episode of unformed, loose stool and to increase fluid intake during diarrhoea episodes. Withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction (see Section 4.2 Dose and Method of Administration).
Skin toxicity.
Rashes, mainly dermatitis acneiform, occurred in patients who received Koselugo (see Section 4.8 Adverse Effects (Undesirable Effects)) and led to dose interruption, dose reduction and permanent discontinuation in some patients.
Other skin toxicities, including severe palmar-plantar erythrodysesthesia syndrome, occurred in an unapproved population of adult patients with multiple tumour types who received Koselugo as a single agent or in combination with other anti-cancer agents.
Monitor for severe skin rashes. Withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction (see Section 4.2 Dose and Method of Administration).
Increased creatine phosphokinase.
Increased creatine phosphokinase (CPK) occurred in patients who received Koselugo (see Section 4.8 Adverse Effects (Undesirable Effects)) and led to dose interruption and dose reduction in some patients. Increased CPK concurrent with myalgia occurred in some patients.
Rhabdomyolysis occurred in an unapproved adult population who received Koselugo as a single agent.
Obtain serum CPK prior to initiating Koselugo, periodically during treatment, and as clinically indicated. If increased CPK occurs, evaluate patients for rhabdomyolysis or other causes. Withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction (see Section 4.2 Dose and Method of Administration).
Embryofetal toxicity.
Based on findings from animal studies and its mechanism of action, Koselugo can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of selumetinib to mice during organogenesis caused reduced fetal weight, adverse structural defects, and effects on embryo-fetal survival at approximate exposures > 3 times the human exposure at the clinical dose of 25 mg/m2 twice daily. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with Koselugo and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with Koselugo and for 1 week after the last dose (see Section 4.6 Fertility, Pregnancy and Lactation).
Risk of choking.
Selumetinib is available as a capsule which must be swallowed whole. Some patients, in particular children < 6 years of age, may be at risk of choking on a capsule formulation due to developmental, anatomical or psychological reasons. Therefore, selumetinib should not be administered to patients who are unable or unwilling to swallow the capsule whole (see Section 4.2 Dose and Method of Administration).
Vitamin E supplementation.
Advise patients not to take any supplemental vitamin E.
Koselugo 10 mg capsules contain 32 mg vitamin E as the excipient, tocofersolan (Australian Approved Name). Koselugo 25 mg capsules contain 36 mg vitamin E as tocofersolan. High doses of vitamin E may increase the risk of bleeding in patients taking concomitant anticoagulant or antiplatelet medications (e.g. warfarin or aspirin). Perform anticoagulant assessments, including international normalised ratio (INR) or prothrombin time (PT) more frequently to detect when the dose adjustments of the anticoagulant or antiplatelet medications are warranted (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Use in the elderly.
The safety and efficacy of Koselugo in adults with NF1-PN older than 65 years of age have not been established. No data are currently available in patients with NF1-PN 65 years of age and older.
Paediatric use.
Safety and effectiveness have been established in paediatric patients 3 years of age and older with NF1 who have inoperable PN and information supporting this use is discussed throughout the Product Information. This indication was expanded to paediatric patients who are 2 years of age and older, because the safety, efficacy and pharmacokinetics of Koselugo in patients who are 2 years of age is expected to be similar to patients at 3 years of age and older.
Effects on laboratory tests.
Please see Section 4.8 Adverse Effects (Undesirable Effects).4.5 Interactions with Other Medicines and Other Forms of Interactions
Pharmacokinetic interactions.
Interaction studies have only been performed in healthy adults (aged ≥ 18 years).
Active substances that may increase selumetinib plasma concentration.
Co-administration with a strong CYP3A4 inhibitor (200 mg itraconazole twice daily for 11 days and 25 mg selumetinib, single oral dose at Day 8) increased selumetinib Cmax by 19% (90% CI 4, 35) and AUC by 49% (90% CI 40, 59) in healthy adult volunteers.
Co-administration with a strong CYP2C19/moderate CYP3A4 inhibitor (400 mg fluconazole single dose at Day 1 followed by 200 mg fluconazole once daily for 10 days and 25 mg selumetinib single oral dose at Day 8) increased selumetinib Cmax by 26% (90% CI 10, 43) and AUC by 53% (90% CI 44, 63) in healthy adult volunteers.
Concomitant use of erythromycin (moderate CYP3A4 inhibitor) or fluoxetine (moderate CYP2C19/strong CYP2D6 inhibitor) is predicted to increase selumetinib AUC by ~30-40% and Cmax by ~20%.
Co-administration with strong inhibitors of CYP3A4 (e.g. clarithromycin, grapefruit juice, oral ketoconazole) or CYP2C19 (e.g. ticlopidine) should be avoided. Co-administration with moderate inhibitors of CYP3A4 (e.g. erythromycin and fluconazole) or CYP2C19 (e.g. omeprazole) should be avoided. If co-administration is unavoidable, patients should be carefully monitored for adverse events and the selumetinib dose should be reduced (see Section 4.2 Dose and Method of Administration; see Table 4).
Active substances that may decrease selumetinib plasma concentrations.
Co-administration with a strong CYP3A4 inducer (600 mg rifampicin daily for 8 days) decreased selumetinib Cmax by 26% (90% CI -17, -34) and AUC by 51% (90% CI -47, -54).
Avoid concomitant use of strong CYP3A4 inducers (e.g. phenytoin, rifampicin, carbamazepine, St. John's Wort) or moderate CYP3A4 inducers with Koselugo.
Active substances whose plasma concentrations may be altered by selumetinib.
In vitro, selumetinib is an inhibitor of OAT3 and the potential for a clinically relevant effect on the pharmacokinetics of concomitantly administered substrates of OAT3 cannot be excluded (see Section 5.2 Pharmacokinetic Properties).
TPGS is a P-gp inhibitor in vitro and it cannot be excluded that it may cause clinically relevant drug interactions with substrates of P-gp (e.g. digoxin or fexofenadine).
The effect of selumetinib on the exposure of oral contraceptives has not been evaluated. Therefore, use of an additional barrier method should be recommended to women using hormonal contraceptives.
Interactions with minimal clinical impact.
In vitro, selumetinib is not an inhibitor of, CYP2A6, CYP2C8, CYP3A4 or CYP2E1, not an inducer of, CYP1A2 and CYP2B6, and did not cause time-dependent inhibition of CYP2C9, CYP2D6 or CYP3A4/5.
In vitro, selumetinib is a reversible inhibitor of CYP2C9, CYP2B6, CYP2D6, an inhibitor of UGT1A3, UGT1A4, UGT1A6 and UGT1A9, an inducer of CYP3A4 and a time-dependent inhibitor of CYP1A2 and CYP2C19; however these effects are not expected to be clinically relevant.
Interactions with transport proteins.
Based on in vitro studies, selumetinib is a substrate for breast cancer resistance protein (BCRP) and P-glycoprotein (P-gp) transporters but is unlikely to be subjected to clinically relevant drug interactions at the recommended paediatric dose. Based on in vitro studies selumetinib is not a substrate for, OATP1B1, or OATP1B3 transporters. In vitro, selumetinib is an inhibitor of BRCP, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, MATE1 and MATE2K but does not inhibit P-gp or OCT1. These in vitro inhibitory effects are not expected to be clinically relevant with the exception of OAT3 where a clinically relevant effect on the pharmacokinetics of concomitantly administered substrates of OAT3 cannot be excluded.
Vitamin E.
Koselugo capsules contain vitamin E as the excipient TPGS. Therefore, patients should avoid taking supplemental vitamin E and anticoagulant assessments should be performed more frequently in patients taking concomitant anticoagulant or antiplatelet medications (see Section 4.4 Special Warnings and Precautions for Use).
Effect of food.
In separate clinical studies, in healthy adult subjects and in adult patients with advanced solid malignancies at a dose of 75 mg, co-administration of Koselugo with a high-fat meal resulted in a mean decrease in Cmax of 50% and 62%, respectively, compared to fasting administration. Selumetinib mean AUC was reduced by 16% and 19%, respectively, and the time to reach maximum concentration (tmax) was delayed by approximately 1.5 hours to 3 hours (see Section 4.2 Dose and Method of Administration).
In healthy adult subjects at a dose of 50 mg, co-administration of Koselugo with a low-fat meal resulted in 60% lower Cmax when compared to fasting administration. Selumetinib AUC was reduced by 38%, and tmax was delayed by approximately 0.9 hours (see Section 4.2 Dose and Method of Administration).
In adolescent patients with NF1 and inoperable PN treated with multiple doses of 25 mg/m2 bid, co-administration of selumetinib with a low-fat meal resulted in 24% lower Cmax when compared to fasting administration. Selumetinib AUC was reduced by 8%, and tmax was delayed by approximately 0.57 hours (see Section 4.2 Dose and Method of Administration).
A population pharmacokinetic (PK) analysis including children and adolescent patients with NF1 and inoperable PN, adult patients with advanced solid malignancies and healthy adult subjects taken from 15 studies showed that concomitant administration of a low or high fat meal resulted in a mean decrease in the exposure (AUC) of selumetinib when compared to fasted administration (23.1% and 20.7%, respectively) which was not considered clinically relevant.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data on the effect of Koselugo on human fertility.
In mice, selumetinib did not affect male mating performance after 10 weeks of dosing up to 20 mg/kg twice daily corresponding to approximately 19 times the human clinical exposure based on AUC at the MRHD. In female mice exposed to selumetinib at up to 37.5 mg/kg twice daily (approximately 65 times the human clinical exposure at the MRHD based on AUC), mating performance and fertility were not affected, but the number of live fetuses was slightly reduced at ≥ 12.5 mg/kg twice daily (18 times the human clinical exposure). Following a three-week treatment withdrawal period, no effects were apparent on any parameter. The no observed adverse effect level (NOAEL) for both maternal toxicity and effects on reproductive performance was 2.5 mg/kg twice daily (approximately 4 times the human clinical exposure). The female fertility associated clinical exposure ratios are derived using the nonpregnant female mice exposure data.
(Category D)
There are no data on the use of selumetinib in pregnant women.
In embryofetal development studies in mice, administration of selumetinib during organogenesis-caused a reduction in the number of live fetuses due to an increase in post-implantation loss, a reduction in mean fetal and litter weights, an increase in unossified or incompletely ossified bones, increased occurrence of open eye and cleft palate at dose levels that did not induce significant maternal toxicity. These effects were seen at ≥ 2.5 mg/kg twice daily (> 3 times the clinical exposure at the MRHD based on AUC) and indicate that selumetinib may have potential to cause defects in the fetus in patients.
Administration of selumetinib to pregnant mice from gestation Day 6 through to lactation Day 20 resulted in reduced pup body weights, and fewer pups met the pupil constriction criterion on Day 21 post-partum at 7.5 mg/kg twice daily (approximately 6 times the clinical exposure based on Cmax). The incidence of malformations (prematurely open eye(s) and/ or cleft palate) was increased at all dose levels (0.5 -7.5 mg/kg twice daily). Malformations occurred at maternal exposure 0.6 times the mean clinical exposure at MRHD based on Cmax.
Koselugo is not recommended during pregnancy.
It is recommended that a pregnancy test should be performed on women of childbearing potential prior to initiating treatment.
Advise women of childbearing potential to avoid becoming pregnant while receiving selumetinib. If a female patient or a female partner of a male patient receiving Koselugo becomes pregnant, she should be apprised of the potential hazard to the fetus.
Both male and female patients (of reproductive potential) should be advised to use effective contraception during and for at least 1 week after completion of treatment with Koselugo. It cannot be excluded that selumetinib may reduce the effectiveness of oral contraceptives, therefore women using hormonal contraceptives should be recommended to use an additional barrier method.
Koselugo is not recommended in women of child-bearing potential not using contraception.
Selumetinib and its active metabolite are excreted in the milk of lactating mice. It is not known whether selumetinib, or its metabolites, are excreted in human milk. A risk to the breast-fed infant cannot be excluded, therefore breast-feeding mothers are advised not to breast-feed during treatment with Koselugo.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed. Koselugo may have a minor influence on the ability to drive and use machines. Fatigue, asthenia and visual disturbances have been reported during treatment with selumetinib and patients who experience these symptoms should observe caution when driving or using machines.
4.8 Adverse Effects (Undesirable Effects)
Clinical trials experience.
Because clinical trials are conducted under widely varying conditions, adverse event rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Neurofibromatosis type 1 (NF1) with inoperable plexiform neurofibromas (PN). Paediatrics 2 years of age and older (SPRINT phase II stratum 1).
The safety of Koselugo was evaluated in SPRINT Phase II Stratum 1 (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Eligible patients were 2-18 years of age with NF1 who had inoperable PN that was causing significant morbidity. Patients were excluded for abnormal LVEF, uncontrolled hypertension (blood pressure > the 95th percentile for age, height, and sex), any current or past history of RVO or RPED, intraocular pressure > 21 mmHg (or upper limit of normal adjusted by age), uncontrolled glaucoma, and inability to swallow whole capsules. Patients received Koselugo 25 mg/m2 orally twice daily (n=50). Among these patients, 88% were exposed for 12 months or longer, 70% were exposed for more than 2 years and 56% were exposed for more than 4 years.
Serious adverse reactions occurred in 30% of patients who received Koselugo. Serious adverse reactions that occurred in 2 or more patients were anaemia, hypoxia, diarrhoea, skin infection and fracture.
Permanent discontinuation due to an adverse event occurred in 12% of patients who received Koselugo. Adverse events resulting in permanent discontinuation of Koselugo included increased blood creatine phosphokinase, increased weight, diarrhoea, paronychia, malignant peripheral nerve sheath tumour, acute kidney injury, and skin ulcer.
Dosage interruptions and dose reductions due to adverse events occurred in 86% and 32% of patients who received Koselugo, respectively. Adverse events requiring a dosage interruption or reduction in ≥ 5% of patients were vomiting, nausea, paronychia, influenza-like illness, diarrhoea, pyrexia, fracture, skin infection, abdominal pain and weight gain.
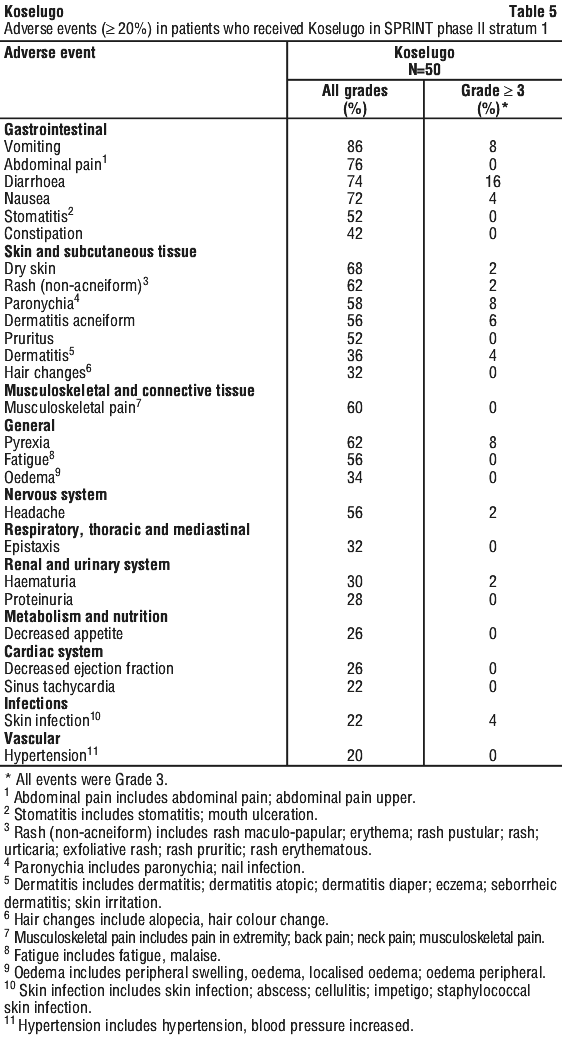
The most common adverse events (≥ 40%) were vomiting, abdominal pain, diarrhoea, nausea, dry skin, pyrexia, rash (non-acneiform), musculoskeletal pain, paronychia, fatigue, dermatitis acneiform, headache, stomatitis, pruritus and constipation.
Table 5 presents the adverse events in SPRINT Phase II Stratum 1.
 Clinically relevant adverse reactions that occurred < 20% of patients include:
Clinically relevant adverse reactions that occurred < 20% of patients include:
Eye: vision blurred.
Gastrointestinal Disorders: dry mouth.
General Disorders: facial oedema, including periorbital oedema and face oedema.
Respiratory, Thoracic and Mediastinal: dyspnoea, including exertional dyspnoea and dyspnoea at rest.
Paediatric pool.
The safety of Koselugo has been evaluated in a pooled safety population of 126 paediatric patients who received a dose ranging from 20 mg/m2 to 30 mg/m2 orally twice daily, from 4 studies in patients with NF1-PN. This paediatric pool includes safety data from SPRINT Phase I (N=24), SPRINT Phase II, Stratum 1 (N=50), China Phase I (N=16), Japan Phase I (N=12), and Phase I Food effect study (N=24). The duration of Koselugo exposure, including dose interruptions, was 12 months or longer (94%), more than 2 years (57%), or more than 3 years (40%).
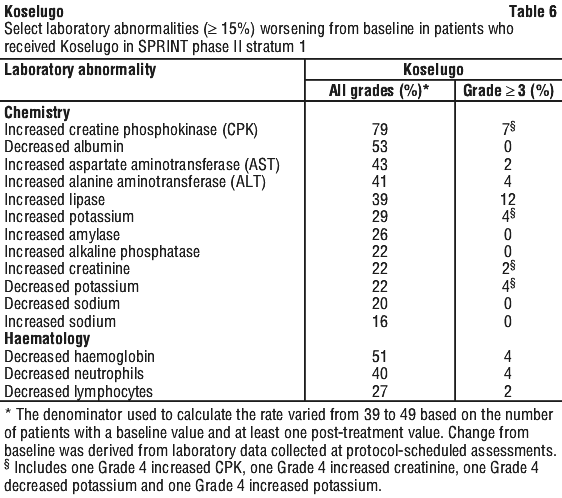
Table 6 presents the laboratory abnormalities in SPRINT Phase II Stratum 1.
Adults ≥ 18 years of age (KOMET phase III).
The safety of Koselugo was evaluated in KOMET (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Eligible patients were 18 years of age or older with NF1 who had symptomatic, inoperable PN. Patients were excluded for abnormal LVEF, uncontrolled hypertension, any current or past history of RVO or RPED/CSR, intraocular pressure > 21 mmHg (or upper limit of normal adjusted by age), uncontrolled glaucoma, and inability to swallow whole capsules. Among the patients (N = 137) who have received Koselugo, the median duration of Koselugo treatment was 11 months with a range of 10 days to 31 months.
Serious adverse events occurred in 13% of patients who received Koselugo. The only serious adverse event to occur in 2 or more patients was cellulitis.
Permanent discontinuation due to an adverse event occurred in 7% of patients who received Koselugo. Adverse events resulting in permanent discontinuation of Koselugo included cellulitis, nausea, dermatitis acneiform, psychiatric decompensation, ulcerative keratitis, and nail disorder.
Dosage interruptions and dose reductions due to adverse events occurred in 31% and 12% of patients who received Koselugo, respectively. Adverse events requiring a dosage reduction in 2 or more patients were paronychia, increased CPK, alopecia, dermatitis acneiform, increased ALT, and increased AST. Adverse events requiring a dosage interruption in 2 or more patients were increased CPK, cellulitis, decreased appetite, headache, abdominal pain, nausea, vomiting, dermatitis acneiform, pyrexia, and ejection fraction decreased.
The most common adverse events (≥ 40%) were rash (all) and rash (acneiform).
Table 7 presents the adverse events in the KOMET study. The 12 cycle (48 weeks) randomisation period for Koselugo versus placebo was followed by a single arm treatment period where all patients received Koselugo (placebo patients crossed over to Koselugo at end of the randomised period). No new adverse events were identified during the open-label period.
 Clinically relevant adverse reactions that occurred < 20% of patients include:
Clinically relevant adverse reactions that occurred < 20% of patients include:
Eye: vision blurred.
Respiratory, Thoracic and Mediastinal: dyspnoea (dyspnoea at rest, exertional dyspnoea).
Gastrointestinal Disorders: stomatitis (aphthous ulcer, gingival swelling, mouth ulceration), constipation, dry mouth.
Skin and Subcutaneous Tissue Disorders: dry skin, hair changes (alopecia, hair colour change), paronychia.
Adult pool.
The pooled safety population of 153 adult patients with NF1 and inoperable PN (25 mg/m2 twice daily, capsules) from 2 studies (NF1-PN adult pool, which includes safety data from the Phase III KOMET study (D134BC00001, N=137) and China Phase I study; adult cohort (D1346C00011, N=16). The median total duration of selumetinib treatment in NF1-PN adult pool was approximately 15 months (range: < 1 - 32 months), 53.6% of patients were exposed to selumetinib treatment for ≥ 12 months and 12% for > 24 months.
Table 8 presents the laboratory abnormalities in the KOMET study.
Adverse drug reactions identified in other clinical trials.
Table 9 presents the adverse drug reactions identified from other clinical trial experience in adult patients (N=347), with multiple tumour types, receiving treatment with selumetinib (75 mg to 100 mg twice daily):

Description of selected adverse reactions.
LVEF reduction.
In the paediatric SPRINT, Phase II Stratum 1 study (N=50), LVEF reduction (PT: ejection fraction decreased) was reported in 13 (26%) patients; all cases were grade 2, asymptomatic and did not lead to discontinuation; one (2%) case led to dose interruption then reduction. Of the 13 patients, 11 patients recovered and for 2 patients the outcome was not reported. The median time to first onset of maximum CTCAE grade LVEF reduction was 232 days (median duration 252 days). The majority of LVEF reduction adverse reactions were reported as reductions from baseline (≥ 10% reduction) but were considered to remain in the normal range.
In the KOMET study (N = 71), LVEF reduction (PT: ejection fraction decreased) was reported in 5 (7%) patients; among them, in 1 (1.4%) patient, the adverse reaction reported was CTCAE grade 3. In 1 (1.4%) patients LVEF decrease led to dose interruption. At the time of analysis, all patients had recovered. The median time to first onset of maximum CTCAE grade LVEF reduction was 121 days (approximately 4 months) [median duration 113 days (approximately 4 months)].
Patients with LVEF lower than the institutional LLN at baseline were not included in the pivotal studies. Decrease in LVEF should be managed using treatment interruption, dose reduction or treatment discontinuation (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Ocular toxicity.
In the paediatric SPRINT, Phase II Stratum 1 study (N=50), grade 1 and 2 adverse reactions of blurred vision were reported in 7 (14%) patients. Two patients required dose interruption. All adverse reactions were managed without dose reduction.
In the KOMET study (N = 71), CTCAE grade 1 event of blurred vision was reported in 3 (4.2%) patients. No patients (0%) required dose interruption. All events were managed without dose reduction and at the time of analysis, all 3 patients had recovered.
If patients report new visual disturbances a complete ophthalmological assessment is recommended. Retinal toxicities can be managed using treatment interruption, dose reduction or treatment discontinuation (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Paronychia.
In the paediatric SPRINT, Phase II Stratum 1 study (N=50), paronychia was reported in 28 (56%) patients, the median time to first onset of maximum grade paronychia adverse reaction was 423 days and the median duration of adverse reactions was 51 days. The majority of these adverse reactions were grade 1 or 2 and were treated with supportive or symptomatic therapy and/or dose modification. Grade ≥ 3 events occurred in 4 (8%) patients. Ten patients (3 with a maximum grade 3 adverse reaction and 7 with a maximum grade 2 adverse reaction) had a selumetinib dose interruption for adverse reactions of paronychia, of whom 5 had dose interruption followed by dose reduction (2 patients required a second dose reduction). In one patient (2%) the event led to discontinuation.
In the KOMET study (N = 71), paronychia was reported in 9 (12.7%) patients. The median time to first onset of maximum CTCAE grade paronychia was 151 days (approximately 5 months) and the median duration of the event was 63 days (approximately 2 months). Seven (9.8%) patients had a maximum CTCAE grade of 1 or 2. Grade 3 events occurred in 2 (2.8%) patients. One patient (1.4%) required dose interruption for adverse event of paronychia, and 2 patients (2.8%) had an event of paronychia that led to dose reduction. Paronychia did not lead to dose discontinuation in any of the patients. At the time of analysis, 7 of the 9 patients had recovered.
Blood creatine phosphokinase (CPK) increase.
In the paediatric SPRINT, Phase II Stratum 1 study (N=50), adverse reactions of blood CPK elevation occurred in 39 (78%) of patients. The median time to first onset of the maximum grade CPK increase was 112 days and the median duration of adverse reactions was 153 days. The majority of adverse reactions were grade 1 or 2 and resolved with no change in selumetinib dose. Grade ≥ 3 adverse reactions occurred in 3 (6%) patients. A grade 4 adverse reaction led to treatment interruption followed by dose reduction.
In the KOMET study (N = 71), adverse events of blood CPK increase occurred in 32 (45.1%) patients. The median time to first onset of the maximum CTCAE grade blood CPK increase was 30.5 days (approximately 1 month), and the median duration of events was 142 days (approximately 5 months). Twenty-seven patients (38%) had maximum CTCAE grade of 1 or 2. A maximum CTCAE grade 3 events occurred in 2 (2.8%) patients, and CTCAE grade 4 events occurred in 3 (4.2%) patients. Three patients had an event of blood CPK increase that led to dose interruptions and dose reduction was required in 2 patients. At the time of analysis, 20 of the 32 patients had recovered.
Gastrointestinal toxicities.
In the paediatric SPRINT, Phase II Stratum 1 study (N=50), vomiting (43 patients, 86%, median duration 3 days), diarrhoea (37 patients, 74%, median duration 6 days), nausea (36 patients, 72%, median duration 15 days), and stomatitis (26 patients, 52%, median duration 27 days) were the most commonly reported gastrointestinal (GI) reactions. The majority of these cases were grade 1 or 2 and did not require any dose interruptions or dose reductions.
Grade 3 adverse reactions were reported for diarrhoea (8 patients, 16%), nausea (2 patients, 4%), and vomiting (4 patients, 8%). For one patient diarrhoea led to dose reduction and subsequent discontinuation. No dose reduction or discontinuation was required for adverse reactions of nausea, vomiting or stomatitis.
In the KOMET study (N = 71), diarrhoea (30 patients, 42.3%), vomiting (18 patients, 25.4%), nausea (18 patients, 25.4%) and stomatitis (13 patients, 18.3%) were the most reported gastrointestinal (GI) events. No CTCAE ≥ Grade 3 events were reported. Dose interruption was required in 2 patients (2.8%) with nausea, and in 1 patient (1.4%) each with diarrhoea and vomiting. Dose reduction occurred in one patient (1.4%) with an ADR stomatitis. One patient reported an event of nausea that led to treatment discontinuation.
Skin toxicities.
In the paediatric SPRINT, Phase II Stratum 1 study (N=50), dermatitis acneiform was observed in 28 (56%) patients (median time to onset 43 days; median duration of 202 days for the maximum CTCAE grade event). The majority of these cases were grade 1 or 2, observed in post-pubertal patients (> 12 years) and did not require any dose interruptions or reductions. Grade 3 adverse reactions were reported in 3 (6%) patients.
Other (non-acneiform) rashes were observed in 27 (54%) patients in the pivotal study and were predominantly grade 1 or 2.
In the KOMET study (N = 71), rashes (acneiform) were observed in 47 (66.2%) patients [median time to onset 13 days; median duration of 156 days (approximately 5 months) for the maximum CTCAE grade event]. Forty-five (63.4%) patients reported AEs with maximum CTCAE grade 1 or 2. CTCAE grade 3 events were reported in 2 (2.8%) patients. In 1 patient (1.4%) acneiform rashes led to dose interruption and dose reduction and in 2 patients (2.8%) led to dose discontinuation. Rashes (non-acneiform) were observed in 17 (23.9%) patients, and all were CTCAE grade 1 or 2.
Hair changes.
In the paediatric SPRINT, Phase II Stratum 1 study (N=50), 16 (32%) of patients experienced hair changes (reported as hair lightening [PT: hair colour changes] in 12 patients (24%) and hair thinning [PT: alopecia] in 12 patients (24%)); in 8 patients (16%) both alopecia and hair colour changes were reported during treatment. All cases were grade 1 and did not require dose interruption or dose reduction.
In the KOMET study (N = 71), 13 (18.3%) patients experienced hair changes [reported as (PT: hair colour changes) in 1 (1.4%) patients and hair thinning (PT: alopecia) in 13 (18%) patients]. All cases were either CTCAE grade 1 or grade 2. No dose interruption was reported and dose reduction in 2 (2.8%) patients.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no specific treatment for overdose. If overdose occurs, patients should be treated supportively with appropriate monitoring as necessary. Dialysis is ineffective in the treatment of overdose.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Selumetinib is an orally available, inhibitor of mitogen-activated protein kinase kinases 1 and 2 (MEK1/2) that is not competitive with respect to ATP. MEK1/2 proteins are critical components of the RAS-regulated RAF-MEK-ERK pathway, which is often activated in different types of cancers. Selumetinib blocks MEK activity and inhibits growth of RAF-MEK-ERK pathway activated cell lines. Therefore, MEK inhibition can block the proliferation and survival of tumour cells in which the RAF-MEK-ERK pathway is activated.
Pharmacodynamics.
In genetically modified mouse models of NF1 that generate neurofibromas that recapitulate the genotype and phenotype of human type 1 neurofibromas, oral dosing of selumetinib inhibits ERK phosphorylation, reduces neurofibroma volume, proliferation, number and growth.
Cardiac electrophysiology.
At a dose 1.5 times the maximum recommended dose, Koselugo does not prolong the QT/QTc interval to any clinically relevant extent.
Clinical trials.
The efficacy of selumetinib in paediatric and adult NF1-PN patients was evaluated in studies as described below.
Paediatric patients 2-18 years of age (SPRINT phase II stratum 1).
The efficacy of Koselugo was evaluated in an open-label, multi-centre, single-arm study [SPRINT Phase II Stratum 1 (NCT01362803)] of 50 paediatric patients with NF1 inoperable PN that caused significant morbidity. Inoperable PN was defined as a PN that could not be surgically completely removed without risk for substantial morbidity due to encasement of, or close proximity to, vital structures, invasiveness, or high vascularity of the PN. Patients received 25 mg/m2 body surface area (BSA) twice daily, for 28 days (1 treatment cycle), on a continuous dosing schedule. Treatment was discontinued if a patient was no longer deriving clinical benefit, experienced unacceptable toxicity or PN progression, or at the discretion of the investigator.
The target PN, the PN that caused relevant clinical symptoms or complications (PN-related morbidities), was evaluated for response rate using centrally read volumetric magnetic resonance imaging (MRI) analysis per Response Evaluation in Neurofibromatosis and Schwannomatosis (REiNS) criteria. Tumour response was evaluated at baseline and while on treatment after every 4 cycles for 2 years, and then every 6 cycles.
Patients had target PN MRI volumetric evaluations and clinical outcome assessments, which included functional assessments and patient reported outcomes.
At enrolment, the median age of the patients was 10.2 years (range: 3.5 - 17.4 years), 60% were male, 84% were Caucasian.
The NF1-PN disease characteristics at baseline are provided in Table 10.
 The primary efficacy endpoint was Objective Response Rate (ORR), defined as the percentage of patients with complete response (defined as disappearance of the target PN) or confirmed partial response (defined as ≥ 20% reduction in PN volume, confirmed at a subsequent tumour assessment within 3-6 months), based on National Cancer Institute (NCI) centralised review. Duration of Response (DoR) was also evaluated.
The primary efficacy endpoint was Objective Response Rate (ORR), defined as the percentage of patients with complete response (defined as disappearance of the target PN) or confirmed partial response (defined as ≥ 20% reduction in PN volume, confirmed at a subsequent tumour assessment within 3-6 months), based on National Cancer Institute (NCI) centralised review. Duration of Response (DoR) was also evaluated.
Efficacy results are provided based on a data cut-off of March 2021, unless stated otherwise.
The primary endpoint, ORR was 68% (95% CI, 53.3 - 80.5). Time to onset of response for the majority of patients (24/34 [70.6%]) was within 8 cycles (range 4 - 42 cycles). The median time to onset of response was 7.2 months (range 3.3 months to 3.2 years).
The median DoR from onset of response was not reached; at the time of data cut-off the median follow-up time was 41.3 months from first dose. Of the 34 patients who had confirmed partial responses, 31 (91.2%) remained in response after 12 months; 26 (76.5%) remained in response after 24 months and 21 (61.8%) remained in response after 36 months. The probability to remain in response after 12, 24 and 36 months, estimated using the Kaplan-Meier method, was 100% (95% CI not estimated), 90.0% (95% CI 72.1 - 96.7) and 86.3% (95% CI 67.3 - 94.6), respectively. The median time from treatment initiation to disease progression while on treatment was not reached. See Table 11.
 At the time of data cut-off or last scan on treatment for patients who had discontinued treatment, 25 (50%) patients remained in confirmed partial response, 1 (2%) had unconfirmed partial responses, 12 (24%) had stable disease and 10 (20%) had progressive disease.
At the time of data cut-off or last scan on treatment for patients who had discontinued treatment, 25 (50%) patients remained in confirmed partial response, 1 (2%) had unconfirmed partial responses, 12 (24%) had stable disease and 10 (20%) had progressive disease.
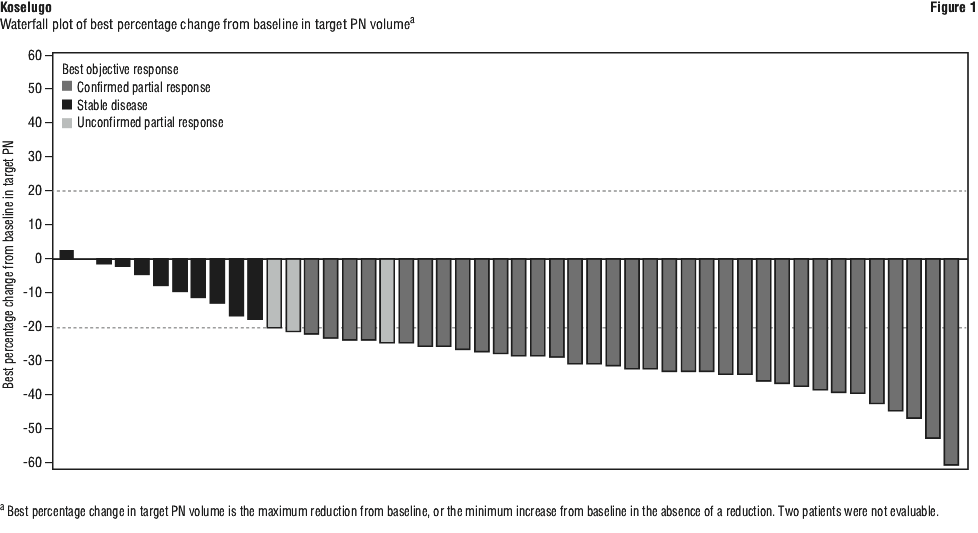
The median best percentage change in PN volume from baseline was -27.85% (range: -60.3% to 2.2%). Figure 1 shows the best percentage change in target PN volume for each patient.
 An independent centralised review of tumour response per REiNS criteria (data cut-off June 2018) resulted in an ORR of 44% (95% CI: 30.0, 58.7).
An independent centralised review of tumour response per REiNS criteria (data cut-off June 2018) resulted in an ORR of 44% (95% CI: 30.0, 58.7).
Adult patients ≥ 18 years of age (KOMET).
The efficacy of Koselugo in adult patients was evaluated in KOMET, a Phase III, randomised, multicentre, double-blind, placebo-controlled trial (NCT04924608). A total of 145 patients were randomised (1:1) to Koselugo 25 mg/m2 (BSA) or placebo twice daily for 12 cycles (28-day cycles). After the end of Cycle 12, or earlier if disease progression was confirmed by the Independent Central Review (ICR), patients initially randomised to placebo crossed over to receive Koselugo in the open-label treatment phase. Treatment was discontinued if a patient was no longer deriving clinical benefit, experienced unacceptable toxicity, patient decision, PN progression, or at the discretion of the investigator.
KOMET enrolled male and female adult (≥ 18 years of age at enrolment) patients with a diagnosis of NF1 who have symptomatic, inoperable PN; at least one target PN measurable by volumetric MRI analysis; chronic target PN pain score documented for a minimum period (for at least 4 days out of 7 days for at least 2 weeks during the screening period); stable chronic PN pain medication use at time of enrolment.
The target PN, the clinically most relevant PN, which is measurable by volumetric MRI analysis and, if relevant, one additional non target PN, was evaluated for response rate using centrally read volumetric MRI analysis per REiNS criteria. Tumour response was evaluated at baseline and while on treatment after every 4 cycles for 2 years, and then every 6 cycles. Patients had target and non target PN MRI volumetric evaluations and clinical outcome assessments.
Demographics and baseline disease characteristics were generally well balanced between the selumetinib and placebo treatment arms. Baseline demographics in the selumetinib and placebo groups were as follows: median age at enrolment was 29 years (range 18 to 60 years), male (51.7%), White (55.9%) and Asian (31%). The NF1-PN disease characteristics at baseline are provided in Table 12.
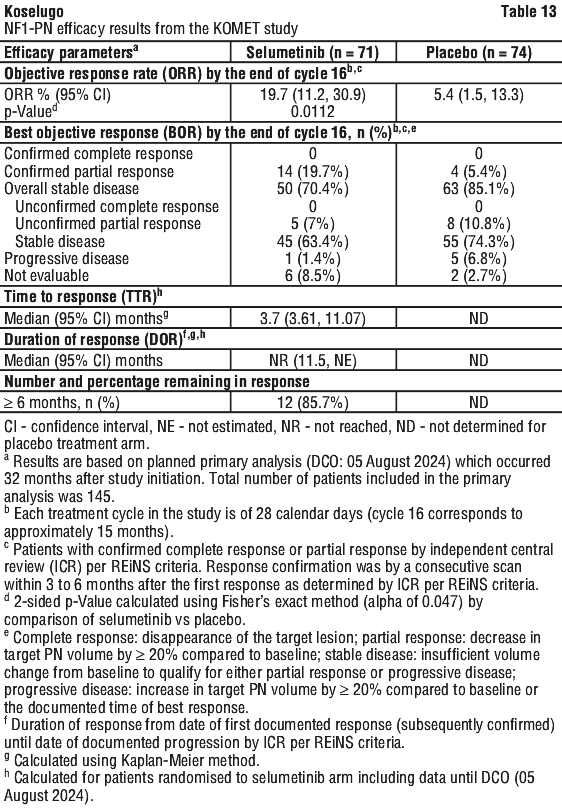
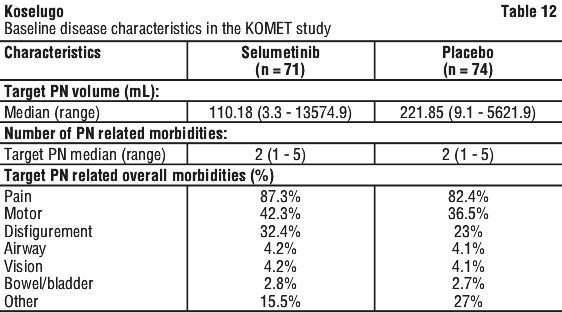 The primary efficacy endpoint was the ORR for selumetinib by the end of Cycle 16. ORR was defined as the percentage of patients with confirmed complete response (disappearance of the target PN, confirmed by a consecutive scan within 3-6 months after the first response) or confirmed partial response (target PN volume decrease ≥ 20%, compared to baseline, confirmed by a consecutive scan within 3-6 months after the first response) by the end of Cycle 16 as determined by ICR per REiNS criteria.
The primary efficacy endpoint was the ORR for selumetinib by the end of Cycle 16. ORR was defined as the percentage of patients with confirmed complete response (disappearance of the target PN, confirmed by a consecutive scan within 3-6 months after the first response) or confirmed partial response (target PN volume decrease ≥ 20%, compared to baseline, confirmed by a consecutive scan within 3-6 months after the first response) by the end of Cycle 16 as determined by ICR per REiNS criteria.
At the planned primary analysis, the study met its primary endpoint demonstrating a statistically significant and clinically meaningful ORR versus placebo. At the time of DCO, the median total duration of exposure was 554 days (approximately 18.2 months) in patients randomised to selumetinib. Efficacy results are presented in Table 13.
Clinical outcome assessments - patient reported outcomes.
A statistically significant difference was not observed between the selumetinib and placebo groups for the key secondary endpoint, the mean change from baseline at Cycle 12 in PAINS-pNF chronic target PN pain intensity score (p = 0.070). The reduction observed in the selumetinib group at Cycle 12 (-2.0; versus -1.3 for placebo) was however considered clinically meaningful.
5.2 Pharmacokinetic Properties
The PK parameters in paediatric patients (3 to ≤ 18 years old) with NF1-PN and in adult patients (≥ 18 years old) with NF1-PN are comparable.
At the recommended dosage of 25 mg/m2 twice daily in paediatric patients (2 to ≤ 18 years old), the mean maximum plasma concentration (Cmax) (coefficient of variation [CV%]) following the first dose and at steady state was 731 (62%) nanogram/mL and 798 (52%) nanogram/mL, respectively. The mean area under the plasma drug concentration curve (AUC0-12h) following the first dose was 2009 (35%) nanogram.h/mL and the AUC0-6h at steady state was 1958 (41%) nanogram.h/mL. Selumetinib AUC and Cmax increase proportionally over a dose range from 20 mg/m2 to 30 mg/m2 (0.8 to 1.2 times the recommended dose). The accumulation was ~1.1 fold following administration of Koselugo 25 mg/m2 twice daily.
In paediatric patients, at a dose level of 25 mg/m2, selumetinib has an apparent oral clearance of 8.8 L/h, mean apparent volume of distribution at steady state of 78 L and mean elimination half-life of ~6.2 hours.
In adult patients (≥ 18 years old), at a dose level of 25 mg/m2, selumetinib has an apparent oral clearance of 14.1 L/h, mean apparent volume of distribution at steady state of 126.1 L and mean elimination half-life of ~9.0 hours.
In the KOMET study at the recommended dosage of 25 mg/m2 twice daily in adult patients (≥ 18 years old), the geometric mean (geometric coefficient of variation [CV%]) maximum plasma concentration (Cmax) was 789 (47%) nanogram/mL, and the mean area under the plasma drug concentration curve (AUC0-12h) was 2986 (43%) nanogram.h/mL at steady state.
Across all age ranges the accumulation range was 1.2-1.5 fold following administration of Koselugo.
Absorption.
In healthy adult subjects, the mean absolute oral bioavailability of selumetinib was 62%.
Following oral dosing, selumetinib is rapidly absorbed, producing peak steady state plasma concentrations (tmax) between 1-1.5 hours post-dose.
Effect of gastric acid reducing agents on selumetinib.
Selumetinib capsules do not exhibit pH dependent dissolution. Koselugo can be used concomitantly with gastric pH modifying agents (i.e. H2-receptor antagonists and proton pump inhibitors) without restrictions, except for omeprazole which is a CYP2C19 inhibitor.
Distribution.
The mean apparent volume of distribution at steady state (Vss) of selumetinib across 20 to 30 mg/m2 (0.8 to 1.2 times the recommended dosage) ranged from 78 L to 171 L based on noncompartmental analysis (NCA) in paediatric patients. In adult patients with a dose of 25 mg/m2, apparent Vss (applying NCA) ranged from 40 to 3710 L indicating moderate distribution into tissue.
In vitro plasma protein binding is 98.4% in humans. Selumetinib mostly binds to serum albumin (96.1%) than α-1 acid glycoprotein (< 35%).
Metabolism.
In vitro, selumetinib undergoes Phase 1 metabolic reactions including oxidation of the side chain, N-demethylation, and loss of the side chain to form amide and acid metabolites. CYP3A4 is the predominant isoform responsible for selumetinib oxidative metabolism with CYP2C19, CYP1A2, CYP2C9, CYP2E1 and CYP3A5 involved to a lesser extent. In vitro studies indicate that selumetinib also undergoes direct Phase 2 metabolic reactions to form glucuronide conjugates principally involving the enzymes UGT1A1 and UGT1A3. Glucuronidation is a significant route of elimination for selumetinib Phase 1 metabolites involving several UGT isoforms.
Following oral dosing of 14C-selumetinib to healthy male subjects, unchanged selumetinib (~40% of the radioactivity) with other metabolites including glucuronide of imidazoindazole metabolite (M2; 22%), selumetinib glucuronide (M4; 7%), N-desmethyl selumetinib (M8; 3%), and N-desmethyl carboxylic acid (M11; 4%) accounted for the majority of the circulating radioactivity in human plasma. The active metabolite, N-desmethyl selumetinib represents less than 10% of selumetinib levels in human plasma but is approximately 3 to 5 times more potent than the parent compound, contributing to about 21% to 35% of the overall pharmacologic activity. N-desmethyl selumetinib is mainly generated by CYP2C19 and catabolised by CYP3A4.
Excretion.
In healthy adult volunteers, following a single oral 75 mg dose of radiolabelled selumetinib, 59% of the dose was recovered in faeces (19% unchanged) while 33% of the administered dose (< 1% as parent) was found in urine by 9 days of sample collection.
Special populations.
Renal impairment.
The exposure of 50 mg oral selumetinib was investigated in adult subjects with normal renal function (n=11) and subjects with ESRD (n=12). The ESRD group showed 16% and 28% lower Cmax and AUC, respectively, with the fraction of unbound selumetinib being 35% higher in ESRD subjects. As a result, the unbound Cmax and AUC ratios were 0.97 and 1.13 in the ESRD group when compared to the group with normal renal function. A small increase, approximately 20% AUC, in the N-desmethyl metabolite to parent ratio was detected in the ESRD group when compared to the normal group. As exposure in ESRD subjects was similar to those with normal renal function, investigations in mild, moderate and severe renally impaired subjects were not performed. Renal impairment is expected to have no meaningful influence on the exposure of selumetinib (see Section 4.2 Dose and Method of Administration).
Hepatic impairment.
Adult subjects with normal hepatic function (n=8) and mild hepatic impairment (Child-Pugh A, n=8) were dosed with 50 mg selumetinib, subjects with moderate hepatic impairment (Child-Pugh B, n=8) were administered a 50 or 25 mg dose, and subjects with severe hepatic impairment (Child-Pugh C, n=8) were administered a 20 mg dose. Selumetinib total dose normalised AUC and unbound AUC were 86% and 69% respectively, in mild hepatic impairment patients, compared to the AUC values for subjects with normal hepatic function. Selumetinib exposure (AUC) was higher in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment; the total AUC and unbound AUC values were 159% and 141% (Child-Pugh B) and 157% and 317% (Child-Pugh C), respectively, of subjects with normal hepatic function (see Section 4.2 Dose and Method of Administration).
Ethnicity.
Following a single-dose, selumetinib exposure appears to be higher in Japanese, non-Japanese-Asian and Indian healthy adult volunteers compared to Western adult volunteers. However, there is considerable overlap with Western subjects when corrected for body weight or BSA (see Section 4.2 Dose and Method of Administration).
Other adult patients (> 18 years old).
The PK parameters in adult healthy volunteers and adult patients with advanced solid malignancies, are similar to those in paediatric patients (3 to ≤ 18 years old) with NF1.
In adult patients with solid malignancies, at a single dose of 75 mg selumetinib, geometric mean (%GCV) Cmax and AUC were 1307 (76%) nanogram/mL and 4736 (37%) nanogram.h/mL, respectively. Peak plasma concentrations of selumetinib were achieved 1.5-hour post-dose with a mean elimination half-life of 7.8 hours. Cmax and AUC increased dose proportionally over a 25 mg to 100 mg dose range, and administration of 75 mg selumetinib twice daily resulted in minimal accumulation of ~1.2 fold.
5.3 Preclinical Safety Data
Genotoxicity.
Selumetinib showed no mutagenic or clastogenic potential in in vitro bacterial reverse mutation [Ames] and mammalian mutagenesis [tk locus TK] assays, and in an in vitro micronucleus assay (in mouse lymphoma L5178Y cells) but produced an increase in micronucleated immature erythrocytes (chromosome aberrations) in in vivo micronucleus assays in mice at ≥ 121 mg/kg, predominantly via an aneugenic mode of action. The exposure at the No Observed Effect Level (NOEL) of 24 mg/kg was approximately 40 times the clinical exposure at the MRHD based on Cmax. The weight of evidence indicates selumetinib has a low genotoxic potential.
Carcinogenicity.
No evidence of tumourigenicity by selumetinib was observed in a 6-month study in transgenic (Tg.rasH2) mice and in a conventional 2-year study in rats. The highest doses tested (15 mg/kg BID in mice and 2.5 mg/kg once daily in male rats and 1 mg/kg once daily in female rats) yielded exposures 14 (male mice), 21 (female mice), 12 (male rat) and 9 (female rat) times the human clinical exposure at the MRHD based on AUC).6 Pharmaceutical Particulars
6.1 List of Excipients
10 mg hard capsule.
Capsule excipient: tocofersolan.
Capsule shell: hypromellose, carrageenan, potassium chloride, titanium dioxide, carnauba wax and purified water.
Printing ink: shellac, iron oxide black, propylene glycol, strong ammonia solution.
25 mg hard capsule.
Capsule excipient: tocofersolan.
Capsule shell: hypromellose, carrageenan, potassium chloride, titanium dioxide, indigo carmine, iron oxide yellow, purified water, carnauba wax and/or maize starch.
Printing ink: iron oxide red, iron oxide yellow, indigo carmine aluminium lake, carnauba wax, shellac, glyceryl monooleate.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
Store in the original bottle to protect from moisture. Keep the bottle tightly closed.
Do not remove desiccant.
6.5 Nature and Contents of Container
HDPE plastic bottle with child-resistant closure and silica gel desiccant, containing 60 hard capsules.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
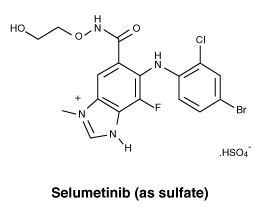
Chemical structure.
Chemical structure of selumetinib (as sulfate).
CAS number.
CAS 943332-08-9 (selumetinib sulfate).7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Summary Table of Changes
