
What is in this leaflet
This leaflet answers some common questions about KOZENIS. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking KOZENIS against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What KOZENIS is used for
KOZENIS contains a medicine called tafenoquine. Malaria is a serious disease spread by infected mosquitoes. KOZENIS is used to eliminate certain forms of malaria (specifically Plasmodium vivax) and help prevent it from coming back. It is given with another medicine (chloroquine) that treats the acute (blood) stage of malaria.
You must get medical advice on which anti-malarial medicines to take. You must ask your doctor or pharmacist if KOZENIS is suitable for the type of malaria to be treated.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
KOZENIS is unlikely to affect your ability to drive or use machines.
The safety and efficacy of tafenoquine have not been established in children and adolescents less than 16 years of age.
Before you take KOZENIS
When you must not take it
Don’t take KOZENIS if you:
- have a condition called glucose-6-phosphate dehydrogenase (G6PD) deficiency (sometimes known as favism)
- do not know your G6PD status or have not had a blood test for G6PD deficiency
- know or think that you are pregnant (see 'Pregnancy and breast-feeding')
- are breastfeeding a baby who is known to have G6PD deficiency or hasn’t been tested for it (see 'Pregnancy and breast-feeding')
- have ever had an allergic reaction to tafenoquine or to a similar medication called primaquine or to one of the ingredients in the tafenoquine tablet, listed at the end of this leaflet.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
Pregnancy and breast-feeding
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding.
Don’t take KOZENIS if you are pregnant.
- Tell your doctor if you are pregnant or planning to become pregnant.
- You should avoid becoming pregnant for at least 3 months after taking KOZENIS.
- If you do become pregnant within 3 months after taking with a dose of KOZENIS, tell your doctor.
Don’t take KOZENIS if you are breastfeeding a baby who has G6PD deficiency or has not been tested for it.
It is not known whether the ingredients of KOZENIS can pass into breast milk. Check with your doctor if you plan to breast-feed, even if you know your child is not G6PD deficient.
Before you start to take it
Because KOZENIS can cause a breakdown of red blood cells in the bloodstream (haemolysis) in persons with G6PD deficiency, a screening test must be performed before you take KOZENIS to see if you have such a deficiency.
After you have taken KOZENIS, contact your doctor if you develop symptoms of haemolytic anaemia (a condition in which red blood cells are destroyed and removed from the bloodstream before their normal lifespan is over). Symptoms of haemolytic anaemia include darkening of the urine, dizziness, confusion, tiredness, or shortness of breath.
KOZENIS stays in your body for up to 3 months. Contact your doctor if adverse reactions occur during this time.
Tafenoquine may cause changes in the haemoglobin in your red blood cells (methaemoglobinemia). Tell your doctor if you been previously told that you have been diagnosed with methaemoglobinia or a related enzyme deficiency.
Tell your doctor if you have currently or previously experienced serious psychiatric disorders such as depression or psychosis.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
If you have not told your doctor about any of the above, tell him/ her before you start taking KOZENIS.
Taking other medicines
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Some medicines and KOZENIS may interfere with each other. These include:
metformin, to treat diabetes
Some medicines may be affected by KOZENIS or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take KOZENIS
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
Take two 150 mg tablets of KOZENIS together as a single dose.
How to take it
KOZENIS is given on the first or second day of a course of chloroquine, which is another medicine for malaria.
When to take it
KOZENIS should be taken with a meal to make sure the right amount of medicine is absorbed.
Do not break or crush the tablets.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much KOZENIS. Do this even if there are no signs of discomfort or poisoning.
While you are using KOZENIS
Things you must do
Tell any other doctors, dentists, and pharmacists who treat you if you have taken this medicine in the past 3 months.
If you are going to have surgery, tell the surgeon or if you have taken this medicine in the past 3 months. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor if you have taken this medicine in the past 3 months. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not take KOZENIS to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well after taking KOZENIS.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Conditions you need to look out for
- Red blood cell breakdown (haemolytic anaemia).
Signs or symptoms may include:
- darkening of the urine
- dizziness or confusion
- tiredness, or shortness of breath.
Get medical help immediately if you get these symptoms. - Severe allergic reactions.
These are very rare in people taking KOZENIS. Signs include:
- raised and itchy rash (hives)
- swelling, sometimes of the face or mouth (angioedema), causing difficulty in breathing
- collapse or loss of consciousness.
Get medical help immediately if you get these symptoms. - Abnormal haemoglobin in the blood (methaemoglobinaemia).
Signs or symptoms may include:
- bluish colouring of the skin, typically starting with the lips
- darkening of the urine
- dizziness
- confusion
- tiredness or shortness of breath
Get medical help immediately if you get these symptoms.
Common side effects
These may affect up to 1 in 10 people:
- Difficulty sleeping
- Headache
- Dizziness
- Nausea
- Vomiting
Common side effects that may show up in blood tests:
- Decreased haemoglobin
- Increased blood methaemoglobin
- Increased liver enzymes
- Increased blood creatinine
Uncommon side effects
These may affect up to 1 in 100 people:
- Anxiety
- Feeling drowsy
- Uncomfortable sensitivity to light
- Effects on the part of your eye called the cornea which may cause changes in your vision
Rare side effects
These may affect up to 1 in 1,000 people:
- Abnormal dreams
Tell your doctor or pharmacist if any of the side effects listed becomes severe or troublesome, or if you notice any side effects not listed in this leaflet.
After using KOZENIS
Storage
Keep your tablets in the pack until it is time to take them.
Do not store KOZENIS or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you not to take this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
The 150 mg film-coated tablets are pink, capsule shaped marked with 'GS J11' on one side.
Ingredients
KOZENIS contains 150 mg of Tafenoquine as the active ingredient.
- Microcrystalline Cellulose
- Mannitol
- Magnesium Stearate
- Purified Water
- Hypromellose
- Titanium Dioxide
- Iron Oxide Red
- Polyethylene Glycol
Supplier
KOZENIS is supplied in Australia by:
GlaxoSmithKline Australia Pty Ltd
Level 4, 436 Johnston Street
Abbotsford Victoria 3067
Australia.
This leaflet was prepared in November 2019.
Trade marks are owned by or licensed to the GSK group of companies
©2019 GSK group of companies or its licensor
Version 3.0
Published by MIMS September 2020
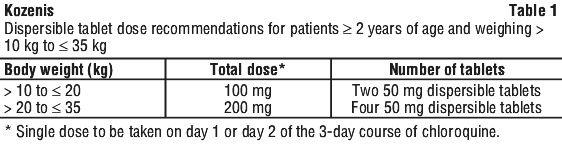 The 50 mg dispersible tablet(s) should be fully dissolved before swallowing. Only water should be used for dissolution. The amount of water for dissolution will depend on the number of tablets prescribed (see the Instructions for Use leaflet in the 50 mg dispersible tablet packaging for complete administration instructions with illustrations). Read the Instructions for Use before starting the therapy.
The 50 mg dispersible tablet(s) should be fully dissolved before swallowing. Only water should be used for dissolution. The amount of water for dissolution will depend on the number of tablets prescribed (see the Instructions for Use leaflet in the 50 mg dispersible tablet packaging for complete administration instructions with illustrations). Read the Instructions for Use before starting the therapy.



 However, no children < 2 years of age and weighing ≤ 10 kg could be recruited in the trial; therefore the use tafenoquine in this age group is not recommended.
However, no children < 2 years of age and weighing ≤ 10 kg could be recruited in the trial; therefore the use tafenoquine in this age group is not recommended. Molecular Formula: C24H28F3N3O3.C4H6O4.
Molecular Formula: C24H28F3N3O3.C4H6O4.