

What is in this leaflet
This leaflet answers some common questions about Leflunomide Sandoz. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Leflunomide Sandoz against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Leflunomide Sandoz is used for
Leflunomide Sandoz is a type of medicine used to treat rheumatoid or psoriatic arthritis.
It belongs to a group of medicines called disease-modifying antirheumatic drugs (DMARDs).
Leflunomide Sandoz helps to slow down the process of joint damage and to relieve the symptoms of the disease, such as joint tenderness and swelling, pain and morning stiffness.
Leflunomide Sandoz works by selectively interfering with the ability of white blood cells called lymphocytes to produce the disease response that ultimately leads to pain, inflammation and joint damage.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children under the age of 18 years.
Before you take Leflunomide Sandoz
When you must not take it
Do not take Leflunomide Sandoz if you have an allergy to:
- leflunomide or teriflunomide
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine if you have or have had any of the following medical conditions:
- any diseases which reduce your body's natural defences, such as bacterial or viral infections
- an illness which severely lowers your body's resistance to disease, such as AIDS
- significant disease of the blood or bone marrow, such as anaemia
- serious allergic skin conditions, such as Stevens-Johnson syndrome, toxic epidermal necrolysis or erythema multiforme
- liver disease
- a condition called hypoproteinaemia (when you do not have enough protein in your blood).
Do not take this medicine if you are pregnant or plan to become pregnant. It may affect your developing baby if you take it during pregnancy. It may increase the risk of birth defects.
You must not become pregnant while taking Leflunomide Sandoz and for a certain period of time after stopping Leflunomide Sandoz. If there is any delay in the onset of menses or you suspect you are pregnant, notify your doctor immediately to test for pregnancy.
Women of childbearing potential must use reliable contraception while taking Leflunomide Sandoz and for a certain period of time after you have stopped taking it.
Do not breast-feed if you are taking this medicine. The active ingredient in Leflunomide Sandoz passes into breast milk and there is a possibility that your baby may be affected.
Do not give this medicine to a child under the age of 18 years. Safety and effectiveness in children younger than 18 years have not been established.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor immediately if you think you could be pregnant while taking Leflunomide Sandoz.
Tell your doctor if you intend to become pregnant or father a child. Leflunomide Sandoz may increase the risk of birth defects. To reduce any risk to the developing baby, you will need to stop taking Leflunomide Sandoz and may need to undergo a wash-out procedure. Your doctor will discuss the washout procedure with you.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding.
Tell your doctor if you have or have had any of the following medical conditions:
- a decrease in the number of white blood cells
- liver problems
- kidney problems
- chronic (ongoing) infections
- an illness which lowered your body's resistance to disease
- peripheral neuropathy
- tuberculosis
- diabetes
- are taking neurotoxic agents
- a history or have a family history of lung problems, such as interstitial lung disease (an inflammation of lung tissue) which is a serious and potentially fatal disease.
Tell your doctor if you plan to have surgery.
Tell your doctor if you have recently been vaccinated or if you need to have a vaccination during treatment with this medicine or for 6 months after stopping Leflunomide Sandoz.
Live vaccines should be avoided while taking this medicine.
If you have not told your doctor about any of the above, tell them before you start taking Leflunomide Sandoz.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Leflunomide Sandoz may interfere with each other. These include:
- teriflunomide, a medicine similar to leflunomide
- warfarin, a medicine used to stop blood clots
- amiodarone, batroxobin, or captopril
- duloxetine, used to treat depression
- theophylline, used to treat respiratory conditions such as asthma
- furosemide, a diuretic
- zidovudine, an anti-retroviral
- some medicines used for diabetes (e.g. tolbutamide, repaglinide, nateglinide, pioglitazone or rosiglitazone)
- some medicines used to treat epilepsy (e.g. phenytoin)
- antibiotics such as rifampicin, cefaclor, benzylpenicillin and ciprofloxacin
- medicines used in cancer treatment such as paclitaxel, topotecan, daunorubicin and doxorubicin
- some medicines used to treat cholesterol such as colestyramine and statins (e.g. rosuvastatin, simvastatin, atorvastatin or pravastatin)
- NSAIDs (non-steroidal anti-inflammatory drugs), such as ibuprofen, naproxen, diclofenac, indomethacin or ketoprofen
- methotrexate and sulfasalazine, used to treat autoimmune disorders such as rheumatoid arthritis
- some types of oral contraceptives
- medicines which have side effects on the blood
- alosetron, used to treat irritable bowel syndrome
- tizanidine, used as a muscle relaxant
- cimetidine, used to treat heart burn and reflux
- some medicines used for tuberculosis (TB).
These medicines may be affected by Leflunomide Sandoz or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines. Your doctor or pharmacist will advise you.
In certain situations, for example, if you experience a serious side effect, you change your medication or you want to fall pregnant, your doctor will ask you to take medication that will help your body get rid of Leflunomide Sandoz faster.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take Leflunomide Sandoz
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the bottle, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how many tablets you will need to take each day. This depends on your condition and whether or not you are taking any other medicines.
The standard dose for this medicine is one 100 mg tablet per day for the first 3 days, and after that one 10 mg or 20 mg tablet daily.
Your doctor may have prescribed a different dose.
Ask your doctor or pharmacist if you are unsure of the correct dose for you. They will tell you exactly how much to take.
How to take it
Swallow the tablets whole with a full glass of water.
When to take it
It does not matter if you take this medicine before or after food.
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
If you are not sure when to take it, ask your doctor or pharmacist.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
Ask your doctor or pharmacist if you are not sure how long to take the medicine for.
If you forget to take it
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
If there is still a long time to go before your next dose, take it as soon as you remember, and then go back to taking it as you would normally.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Leflunomide Sandoz. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include diarrhoea, stomach pain, changes in blood, or liver damage.
While you are taking Leflunomide Sandoz
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Leflunomide Sandoz.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately. This medicine may cause serious birth defects.
Tell your doctor before stopping contraception. You must continue using appropriate reliable contraception (the 'Pill' or condoms) while you are taking Leflunomide Sandoz. If you plan to stop your contraception, you must first discuss this with your doctor.
If you have an infection or notice a fever or signs of an infection while taking this medicine, tell your doctor immediately.
Tell your doctor if you develop symptoms such as pins and needles or tingling in the hands or feet or numbness or weakness of the arms and legs, or become clumsy, have visual problems and problems speaking.
If your skin becomes itchy or yellow, if the whites of your eyes become yellow, or if you start to bleed or bruise easily, stop taking it and tell your doctor or pharmacist immediately. You may be developing a liver problem. Your doctor may need to take blood samples to monitor the health of your liver and blood cells while you are taking Leflunomide Sandoz.
Tell your doctor immediately if you develop new or worsening symptoms such as a cough or trouble breathing. Inflammation of the lung tissue which can be fatal, has been reported in some patients.
Tell your doctor immediately and stop taking your medicine if you develop any symptoms of liver problems, including yellowing of eyes, itchy and yellowing skin, bruising and bleeding easily. Your doctor will check the health of your liver using blood tests on a regular basis while you are taking Leflunomide Sandoz.
Tell your doctor if you need to have a vaccination during treatment with this medicine or for 6 months after stopping treatment.
Tell your doctors if you are going to have surgery or an anaesthetic or are going into hospital.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor will do blood tests and monitor your blood pressure before starting and during treatment. This is to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not take more than the recommended dose unless your doctor tells you to.
Do not take Leflunomide Sandoz to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how Leflunomide Sandoz affects you. This medicine may cause tiredness, in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Be careful when drinking alcohol while you are taking this medicine. The effects of alcohol could be made worse while taking Leflunomide Sandoz. It is recommended that you minimise your alcohol intake while taking Leflunomide Sandoz.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Leflunomide Sandoz.
This medicine helps most people with arthritis, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- diarrhoea
- nausea, vomiting, abdominal pain
- rashes, itchy skin
- hair loss
- weight loss
- unusual tiredness or weakness
- joint, muscle and general pain
- varicose veins
- constipation
- dry mouth
- problems sleeping
- headache
- dizziness
- sweating
- blurred vision
- changes in taste
- pins and needles sensation in hands or feet.
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- signs and symptoms of severe
- infection e.g. fever
- severe upper stomach pain, often with nausea and vomiting, anorexia, or diarrhoea
- joint pain, swelling, stiffness, muscle weakness
- problems with tendons in the legs
- severe skin rash, scaling or sores and blisters (sometimes in your mouth including ulcers), often with flu-like symptoms
- your skin becomes pale, you start to feel tired, you become prone to infections or bruising
- if you develop new or worsening symptoms such as cough or trouble breathing, with or without a fever
- blisters and bleeding in the lips, eyes, mouth, nose and genitals
- thickened patches of red skin
- irregular, fast or slow heartbeat
- shortness of breath, tiredness, swelling in the legs, dizziness
- chest pain
- dark stools
- blood in the urine
- excessive thirst, frequent urination or decreased urine
- anxiety, depression, delirium, confusion
- changes to your liver and pancreas or blood, which may be detectable by blood tests
- temporary changes to sperm.
The above list includes serious side effects that may require medical attention. Serious side effects are rare.
Leflunomide Sandoz decreases your body's immune response and can cause some of the side effects listed above.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- swelling of the face, lips, mouth or throat, which may cause difficultly in swallowing or breathing
- hives
- fainting
- yellowing of the skin and eyes (jaundice).
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation. These side effects are very rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After taking Leflunomide Sandoz
Storage
Keep your tablets in the bottle until it is time to take them. If you take the tablets out of the bottle they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store Leflunomide Sandoz or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Return any unused medicine to your pharmacist.
Product description
What it looks like
Tablets, film coated
10 mg: white, round biconvex tablets 30’s (bottle)
20 mg: yellow, round biconvex tablets with a scoreline on one side 30’s (bottle)
Ingredients
Leflunomide Sandoz contains 10 mg or 20 mg of leflunomide as the active ingredient.
The tablets also contain the following inactive ingredients:
- microcrystalline cellulose,
- lactose monohydrate
- maize starch
- povidone
- crospovidone
- colloidal anhydrous silica
- magnesium stearate
- Opadry II complete film coating system OY-LS-28908 White (10 mg only)
- Opadry aqueous film coating OY-SR-6497 Yellow (20 mg only).
Leflunomide Sandoz 10 mg and 20 mg tablets contain sugars (as lactose).
Distributor
Leflunomide Sandoz is distributed in Australia by:
Sandoz Pty Ltd
19 Harris Street
Pyrmont NSW 2009
This leaflet was prepared in September 2020.
10mg tablets: AUST R 210905
20mg tablets: AUST R 210906
Leflunomide Sandoz_cmi\0920/03
Published by MIMS November 2020
 In addition, the following adverse events have been reported in 1 to 3% of the RA patients in the leflunomide treatment group in controlled clinical trials.
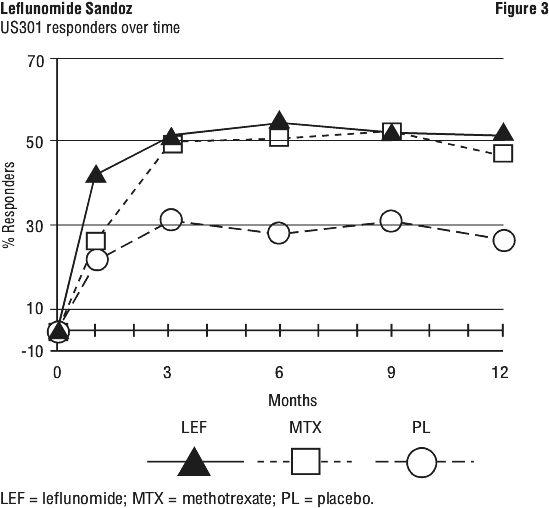
In addition, the following adverse events have been reported in 1 to 3% of the RA patients in the leflunomide treatment group in controlled clinical trials. ACR 20, 50 and 70% response rates in all treatment groups of the US301 trial are shown in Figure 2. In US301, the ACR 20% response rate of patients treated with leflunomide was significantly increased from that seen in placebo treated patients and similar to that seen in patients treated with methotrexate. Similarly, ACR 50% and 70% responses were approximately two times higher in patients treated with leflunomide or methotrexate than in patients who received placebo.
ACR 20, 50 and 70% response rates in all treatment groups of the US301 trial are shown in Figure 2. In US301, the ACR 20% response rate of patients treated with leflunomide was significantly increased from that seen in placebo treated patients and similar to that seen in patients treated with methotrexate. Similarly, ACR 50% and 70% responses were approximately two times higher in patients treated with leflunomide or methotrexate than in patients who received placebo.


 The use of a loading dose may increase toxicity associated with leflunomide when used in children. The population pharmacokinetic data indicate that adult doses of leflunomide would be inappropriate in some children who would require a dose calculated from bodyweight. The safety and effectiveness of leflunomide in paediatric patients have not been fully evaluated. Leflunomide is not recommended for use in patients under 18 years.
The use of a loading dose may increase toxicity associated with leflunomide when used in children. The population pharmacokinetic data indicate that adult doses of leflunomide would be inappropriate in some children who would require a dose calculated from bodyweight. The safety and effectiveness of leflunomide in paediatric patients have not been fully evaluated. Leflunomide is not recommended for use in patients under 18 years. In a differing analysis which excluded some patients, the Psoriasis Area and Severity Index (PASI) was assessed to reflect changes in the extent and severity of psoriasis lesions as judged by erythema, desquamation and infiltration. Leflunomide resulted in a significant improvement in PASI scores over the 24 week study relative to placebo, with a mean (± SD) improvement of 22.4% (± 51.6%) in the leflunomide group compared with a deterioration of 2.2% (± 70.4%) in the placebo group (p = 0.0030). See Table 4.
In a differing analysis which excluded some patients, the Psoriasis Area and Severity Index (PASI) was assessed to reflect changes in the extent and severity of psoriasis lesions as judged by erythema, desquamation and infiltration. Leflunomide resulted in a significant improvement in PASI scores over the 24 week study relative to placebo, with a mean (± SD) improvement of 22.4% (± 51.6%) in the leflunomide group compared with a deterioration of 2.2% (± 70.4%) in the placebo group (p = 0.0030). See Table 4. Compared with the placebo group, a significantly greater proportion of patients in the leflunomide group experienced a greater than or equal to 50% reduction in PASI scores (PASI 50; 18.9 versus 30.4%; p = 0.050) a greater than or equal to 75% reduction in PASI scores (PASI 75; 7.8 versus 17.4%; p = 0.048) from baseline.
Compared with the placebo group, a significantly greater proportion of patients in the leflunomide group experienced a greater than or equal to 50% reduction in PASI scores (PASI 50; 18.9 versus 30.4%; p = 0.050) a greater than or equal to 75% reduction in PASI scores (PASI 75; 7.8 versus 17.4%; p = 0.048) from baseline. Relative to an oral solution, leflunomide tablets are 80% bioavailable. Food does not affect the bioavailability of leflunomide.
Relative to an oral solution, leflunomide tablets are 80% bioavailable. Food does not affect the bioavailability of leflunomide.

