
What is in this leaflet
This leaflet answers some common questions about Levetiracetam-GH. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Levetiracetam-GH against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Levetiracetam-GH is used for
Levetiracetam-GH is used to control epilepsy.
Epilepsy is a condition where you have repeated seizures. There are many different types of seizures, ranging from mild to severe.
This medicine belongs to a group of medicines called antiepileptics. These medicines are thought to work by controlling brain chemicals which send signals to nerves so that seizures do not happen.
Levetiracetam-GH may be used alone, or in combination with other medicines, to treat your condition.
Your doctor may have prescribed this medicine in addition to your current therapy.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
There is no evidence that this medicine is addictive.
This medicine is available only with a doctor’s prescription.
The safety and effectiveness of Levetiracetam-GH has not been established in patients less than 4 years of age.
Before you take Levetiracetam-GH
When you must not take it
Do not take Levetiracetam-GH if you have an allergy to:
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath;
- wheezing or difficulty breathing;
- swelling of the face, lips, tongue or other parts of the body;
- rash, itching or hives on the skin.
Do not take this medicine after the expiry date (eg. EXP) printed on the pack.
Do not take this medicine if the packaging is torn or shows signs of tampering or if the tablets do not look quite right. If this medicine has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor or pharmacist.
Before you start to take it
Tell your doctor or pharmacist if you have allergies to:
- any other medicines, especially barbiturates (such as phenobarbitone) or any other antiepileptic medicines (such as carbamazepine, lamotrigine or valproate);
- any other substances, such as foods, preservatives or dyes.
Tell your doctor if you have or have had any medical conditions, especially the following:
- kidney problems;
- liver problems.
Tell your doctor if you are pregnant or intend to become pregnant. Levetiracetam-GH may affect your developing baby if you take it during pregnancy. However, it is very important to control your seizures while you are pregnant. Your doctor will outline and weigh up all the risks and benefits of taking Levetiracetam during pregnancy to help decide whether or not you should take it.
Tell your doctor if you are breastfeeding or plan to breastfeed. The active ingredient in Levetiracetam-GH passes into breast milk and there is a possibility that your baby may be affected. Your doctor will discuss the risks and benefits of using Levetiracetam-GH if you are breastfeeding.
If you have not told your doctor or pharmacist about any of the above, tell them before you start taking Levetiracetam-GH.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Levetiracetam-GH does not interact with the oral contraceptive pill. However, you may be given Levetiracetam-GH together with other antiepileptic medicines that do interact and may affect the effectiveness of your contraceptive. Your doctor may advise you to use an additional method of contraception if you take Levetiracetam-GH with other antiepileptic medicines.
How to take Levetiracetam-GH
How much to take
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
Your doctor will tell you how much Levetiracetam-GH you will need to take each day. This may depend on your age, your condition and whether or not you are taking any other medicines.
For patients 12 years of age and older, the dosage is generally between 1000 mg and 3000 mg each day, taken in two doses.
For children 4 to 11 years of age the doctor will calculate the dosage based on the child’s weight and tell you how much to give. The medicine is to be given twice daily.
Your doctor may start you on a low dose of Levetiracetam-GH first. Your doctor will slowly increase the amount of medicine until you are taking enough to control your epilepsy and you are not having seizures.
If you do not understand the instructions on the pack, ask your doctor or pharmacist for help.
How to take it
Swallow the tablets whole with a glass of water.
When to take it
Take Levetiracetam-GH twice a day, once in the morning and once at night. Take it, at about the same time each day. Taking your medicine at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it
Most antiepileptic medicines take time to work, so do not be discouraged if you do not feel better straight away.
Continue taking your medicine for as long as your doctor tells you to. This medicine helps control your condition, but does not cure it. Therefore you must take your medicine every day, even if you feel well.
Do not stop taking Levetiracetam-GH, or change the dosage, without checking with your doctor. Do not let yourself run out of medicine over the weekend or on holidays. Stopping Levetiracetam-GH suddenly may cause unwanted side effects or make your condition worse. Your doctor will slowly reduce your dose before you can stop taking it completely.
If you forget to take it
Contact your doctor if you have missed one or more doses.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26 or New Zealand 0800 764 766) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Levetiracetam-GH. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include feeling drowsy.
While you are taking Levetiracetam-GH
Things you must do
Tell your doctor immediately if you notice an increase in seizures.
Tell your doctor immediately if you have symptoms of depression or thoughts of harming yourself.
Tell any other doctors, dentists, and pharmacists who are treating you that you are taking this medicine.
If you are about to be started on any new medicine, tell your doctor, dentist or pharmacist that you are taking Levetiracetam-GH.
Before you have any surgery or emergency treatment, tell your doctor or dentist that you are taking Levetiracetam-GH.
Tell your doctor if you feel Levetiracetam-GH is not helping your condition. Your doctor may need to change your medicine.
Tell your doctor if, for any reason, you have not taken this medicine exactly as prescribed. Otherwise, your doctor may change your treatment unnecessarily.
If you become pregnant while taking this medicine, tell your doctor.
Be sure to keep all of your doctor’s appointments so that your progress can be checked. Your doctor will check your progress and may want to take some tests from time to time. This helps to prevent unwanted side effects.
Things you must not do
Do not give Levetiracetam-GH to anyone else, even if their symptoms seem similar to yours or they have the same condition as you.
Do not take Levetiracetam-GH to treat any other complaints unless your doctor tells you to.
Things to be careful of
Be careful driving or operating machinery until you know how this medicine affects you. Children should be careful doing things like riding bicycles or climbing trees.
As with other antiepileptic medicines, Levetiracetam-GH may cause dizziness or drowsiness in some people.
This is more frequent at the beginning of treatment or after an increase in the dose.
If you are feeling dizzy or drowsy do not drive, operate machinery, or do anything else that could be dangerous. Children should not ride a bike, climb trees or do anything else that could be dangerous if they are feeling dizzy or drowsy.
Be careful when drinking alcohol while taking this medicine. Combining Levetiracetam-GH and alcohol can make you more drowsy.
Your doctor may suggest you avoid alcohol while you are being treated with Levetiracetam-GH.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Levetiracetam-GH.
This medicine helps most people with epilepsy, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
If you get any side effects, do not stop taking Levetiracetam-GH without first talking to your doctor or pharmacist.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- dizziness;
- feeling weak;
- headache;
- common cold;
- upset stomach;
- diarrhoea;
- feeling tired, drowsy or sleepy.
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- mood changes such as depression, nervousness, aggression, anger, anxiety, confusion, hallucination, irritability;
- feelings of depression;
- upper respiratory tract infections;
- weight loss.
The above list includes serious side effects that may require medical attention.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- thoughts of harming yourself;
- more frequent or more severe seizures;
- shortness of breath;
- wheezing or difficulty breathing;
- swelling, blistering or peeling skin around the face, lips, mouth, throat, tongue, genitals or other parts of the body;
- rash, itching or hives on the skin.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed above may happen in some people.
After taking Levetiracetam-GH
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of their pack they will not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store Levetiracetam-GH or any other medicine in the bathroom or near a sink.
Do not leave it on a window sill or in the car on hot days. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Levetiracetam-GH are available in three strengths:
- Levetiracetam-GH 250 mg: Blue, oblong -shaped, biconvex film-coated tablets, debossed with ‘250’ on one side and score line on the other side.
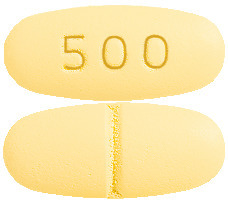
- Levetiracetam-GH 500 mg: Yellow, oblong-shaped, biconvex film-coated tablets, debossed with ‘500’ on one side and score line on the other side.
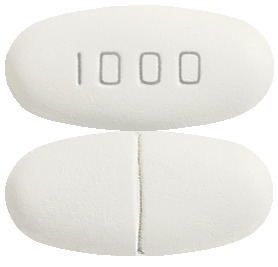
- Levetiracetam-GH 1000 mg: White to off white, oblong -shaped, biconvex film-coated tablets, debossed with ‘1000’ on one side and score line on the other side.
Levetiracetam 250 mg, 500 mg and 1000 mg tablets are packed in blister packs, pack sizes of 60 tablets.
Ingredients
Active ingredient
Each tablet contains 250 mg, 500 mg or 1000 mg of levetiracetam as the active ingredient.
Other ingredients
- maize starch;
- colloidal anhydrous silica;
- croscarmellose sodium;
- povidone;
- microcrystalline cellulose;
- purified talc;
- magnesium stearate.
Levetiracetam-GH are film-coated & the coating for each tablet strength contains:
- polyvinyl alcohol;
- macrogol 6000 and macrogol 3350;
- purified talc; and
- titanium dioxide.
The following strengths also contain:
- Indigo carmine CI73015 (250 mg tablets only);
- Iron oxide yellow CI77492 (500 mg tablets only).
Levetiracetam-GH do not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Australian Registration Numbers
Levetiracetam-GH 250 mg: AUST R 161294.
Levetiracetam-GH 500 mg: AUST R 161295.
Levetiracetam-GH 1000 mg: AUST R 161296.
Distributor
Generic Health Pty Ltd
Suite 2, Level 2
19-23 Prospect Street
Box Hill, VIC, 3128
Australia
Email: [email protected]
Telephone: +61 3 9809 7900
Website: www.generichealth.com.au
This leaflet was prepared in March 2020.
Published by MIMS June 2020

 The ClCr is adjusted for body surface area (BSA) as follows. See Equation 2.
The ClCr is adjusted for body surface area (BSA) as follows. See Equation 2.
 For children with renal impairment, levetiracetam dose needs to be adjusted based on the renal function as levetiracetam clearance is related to renal function. This recommendation is based on a study in adult renally impaired patients.
For children with renal impairment, levetiracetam dose needs to be adjusted based on the renal function as levetiracetam clearance is related to renal function. This recommendation is based on a study in adult renally impaired patients. The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications. In the pooled safety analysis, there was no clear dose response relationship but incidence and severity of CNS related undesirable effects decreased over time.
In the pooled safety analysis, there was no clear dose response relationship but incidence and severity of CNS related undesirable effects decreased over time. The incidence of serious adverse events in placebo controlled studies was 9.9% in the levetiracetam group versus 8.9% in the placebo group. Many of the serious adverse events are typical for a population of patients with epilepsy.
The incidence of serious adverse events in placebo controlled studies was 9.9% in the levetiracetam group versus 8.9% in the placebo group. Many of the serious adverse events are typical for a population of patients with epilepsy. Other events occurring in 2% or more of paediatric patients treated with levetiracetam but as or more frequent in the placebo group were the following: abdominal pain, allergic reaction, ataxia, convulsion, epistaxis, fever, headache, hyperkinesia, infection, insomnia, nausea, otitis media, rash, sinusitis, status epilepticus (not otherwise specified), thinking abnormal, tremor and urinary incontinence.
Other events occurring in 2% or more of paediatric patients treated with levetiracetam but as or more frequent in the placebo group were the following: abdominal pain, allergic reaction, ataxia, convulsion, epistaxis, fever, headache, hyperkinesia, infection, insomnia, nausea, otitis media, rash, sinusitis, status epilepticus (not otherwise specified), thinking abnormal, tremor and urinary incontinence. The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the three treatment groups (x-axis) is presented in Figure 1.
The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the three treatment groups (x-axis) is presented in Figure 1.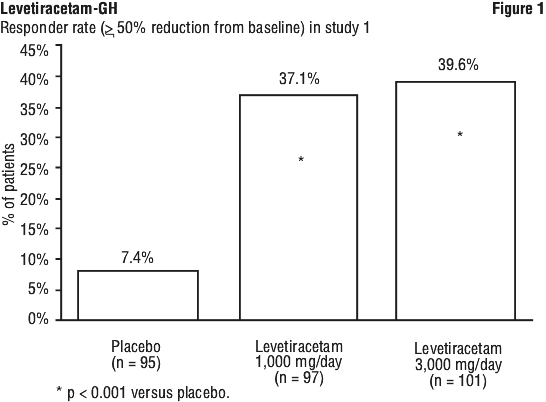
 The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the three treatment groups (x-axis) is presented in Figure 2.
The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the three treatment groups (x-axis) is presented in Figure 2. The comparison of levetiracetam 2000 mg/day to levetiracetam 1000 mg/day for responder rate was statistically significant (p = 0.02). Analysis of the trial as a crossover yielded similar results.
The comparison of levetiracetam 2000 mg/day to levetiracetam 1000 mg/day for responder rate was statistically significant (p = 0.02). Analysis of the trial as a crossover yielded similar results. The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the two treatment groups (x-axis) is presented in Figure 3.
The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the two treatment groups (x-axis) is presented in Figure 3.
 The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the two treatment groups (x-axis) is presented in Figure 4.
The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomised treatment period (titration plus evaluation period) within the two treatment groups (x-axis) is presented in Figure 4.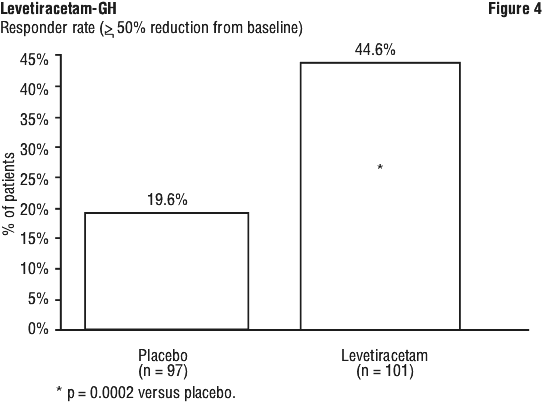

 The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in PGTC seizure frequency over the entire randomised treatment period (titration + evaluation period) within the two treatment groups (x-axis) is presented in Figure 5.
The percentage of patients (y-axis) who achieved ≥ 50% reduction in weekly seizure rates from baseline in PGTC seizure frequency over the entire randomised treatment period (titration + evaluation period) within the two treatment groups (x-axis) is presented in Figure 5. When levetiracetam was used to treat PGTC seizures in adults and adolescents with IGE, there was no effect on the frequency of absences.
When levetiracetam was used to treat PGTC seizures in adults and adolescents with IGE, there was no effect on the frequency of absences. Chemical Name: (S)-alpha-ethyl-2-oxo-1-pyrrolidine acetamide.
Chemical Name: (S)-alpha-ethyl-2-oxo-1-pyrrolidine acetamide.