What is in this leaflet
This leaflet answers some common questions about LORVIQUA. It does not contain all the available information. It does not take the place of you talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking LORVIQUA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What LORVIQUA is used for
LORVIQUA is used to treat a rare type of lung cancer that is caused by defects in a gene called anaplastic lymphoma kinase (ALK).
LORVIQUA blocks the action of an enzyme called ‘ALK tyrosine kinase’. Abnormal forms of this enzyme (due to the fault in the ALK gene) help encourage cancer cell growth.
LORVIQUA may slow down or stop the growth of your cancer. It may also help to shrink your cancer.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
LORVIQUA is available only with a doctor's prescription.
It is not addictive.
There is not enough information to recommend the use of this medicine for children.
Before you take LORVIQUA
When you must not take it
Do not take LORVIQUA if you are allergic to any medicine containing lorlatinib or to any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
LORVIQUA contains lactose, a type of sugar found in milk or dairy products.
Talk to you doctor if you have been told that you cannot tolerate or digest some sugars.
Do not take LORVIQUA if you are taking any of the following medicines:
- rifampicin, a medicine used to treat tuberculosis
- carbamazepine, phenytoin, medicines used to treat epilepsy
- enzalutamide, a medicine used to treat prostate cancer
- mitotane, a medicine used to treat cancer of the adrenal glands
- medicines containing St John's wort (hypericum perforatum, a herbal preparation).
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breast-feed while taking this medicine or for 7 days after the last dose. The active ingredient in LORVIQUA may pass into breast milk and there is a possibility that your baby may be affected.
Do not give this medicine to a child. Safety and effectiveness in children have not been established.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- high levels of blood cholesterol or triglycerides
- heart problems, including reduced heart rate, or if the results of an electrocardiogram (ECG) have shown that you have an abnormality of the electrical activity of your heart known as "prolonged PR interval" or "AV block"
- high levels of the enzymes known as amylase or lipase in the blood or a condition such as pancreatitis that can raise the levels of these enzymes
- cough, chest pains, shortness of breath, or worsening of respiratory symptoms or ever had a lung condition called pneumonitis
- liver problems
- kidney problems
- central nervous system (CNS) effects.
Your doctor should perform blood tests to check the level of cholesterol and triglycerides in your blood before you start treatment with LORVIQUA and regularly during treatment.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
You should avoid falling pregnant while using this medicine.
Use highly effective contraception (e.g. double-barrier contraception such as condom and diaphragm) during treatment and for at least 21 days after stopping treatment.
LORVIQUA may reduce the effectiveness of hormonal contraceptive methods (e.g. birth control pill). If hormonal contraception is unavoidable it must be used with a form of barrier contraception such as a condom.
If your male partner is being treated with LORVIQUA, he must use a condom during treatment and for at least 14 weeks after stopping treatment.
Talk to your doctor about the right methods of contraception for you and your partner.
If you become pregnant when taking LORVIQUA or during the 3 weeks after taking your last dose, tell your doctor straight away.
LORVIQUA may affect male fertility.
Talk to your doctor about fertility preservation before taking LORVIQUA.
If you have not told your doctor about any of the above, tell him/ her before you start taking LORVIQUA.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and LORVIQUA may interfere with each other. These include:
- boceprivir, a medicine used to treat hepatitis C
- medicines used to treat AIDS/ HIV such as efavirenz, cobicistat, ritonavir, paritaprevir in combination with ritonavir and ombitasvir and/or dasabuvir, and ritonavir in combination with either danoprevir, elvitegravir, indinavir, lopinavir, saquinavir or tipranavir
- medicines used to treat fungal infections such as itraconazole, ketoconazole, posaconazole, voriconazole
- troleandomycin, a medicine used to treat certain types of bacterial infections
- quinidine, a medicine used to treat irregular heartbeat and other heart problems
- medicines used to treat severe pain such as alfentanil and fentanyl
- medicines used to prevent organ transplant rejection such as ciclosporin, sirolimus and tacrolimus.
These medicines may be affected by LORVIQUA or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take LORVIQUA
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
The recommended dose is 100 mg taken orally once daily.
Your doctor may lower your dose, stop your treatment for a short time or stop your treatment completely if you feel unwell.
How to take it
Swallow the tablets whole and do not crush, chew or split the tablets.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food. However, you should avoid eating grapefruit or drinking grapefruit juice while taking LORVIQUA.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
It is important to take this medicine every day for as long as your doctor prescribes it for you.
If you forget to take it
If it is less than 4 hours before your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice if you think that you or anyone else may have taken too much LORVIQUA. Do this even if there are no signs of discomfort or poisoning. You may need medical attention.
While you are taking LORVIQUA
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking LORVIQUA.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately.
Tell your doctor immediately if you develop any of the following:
- changes in your heartbeat (fast or slow), light-headedness, fainting, dizziness or shortness of breath.
Your doctor may do some tests (electrocardiograms) to check that there are no problems with your heart during treatment with LORVIQUA. If the results are abnormal he/she may decide to reduce your dose or stop your treatment. - difficulty speaking, including slurred or slow speech
- change in your mood (including depression, euphoria and mood swings), feelings of irritability, aggression, agitation, anxiety or a change in your personality, memory loss or impairment, episodes of confusion
Tell your doctor if you notice any of the following:
- you feel more tired than usual
- your skin and whites of your eyes turn yellow
- your urine turns dark brown (tea colour)
- you have nausea, vomiting or decreased appetite
- you have pain in the upper area of and/or on the right side of your stomach
- you have itching
- you bruise more easily than usual
- you develop a fever
- you develop a cough, chest pains, shortness of breath, or worsening of respiratory symptoms.
Your doctor may do some tests (blood tests, scans) to check your liver function and pancreas. If the results are abnormal he/she may decide to reduce your dose or stop your treatment.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not take LORVIQUA to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
If you are not able to take the medicine as your doctor has prescribed, contact your doctor right away.
Things to be careful of
Be careful driving or operating machinery until you know how LORVIQUA affects you.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking LORVIQUA.
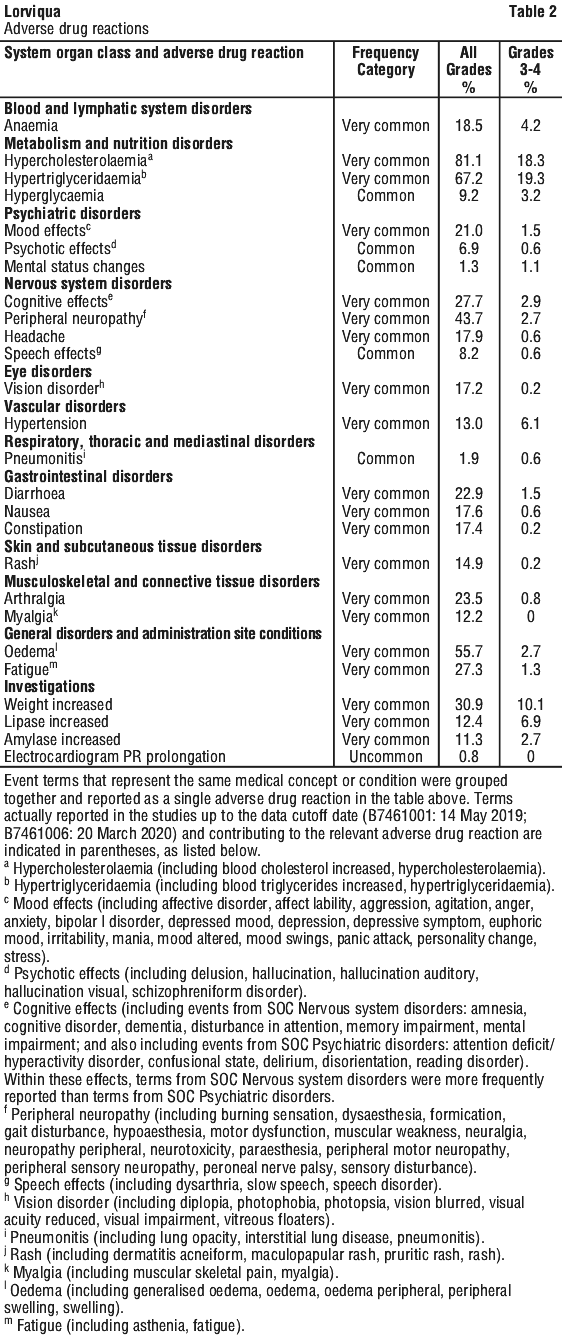
Like all medicines, LORVIQUA can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following:
- effects on memory, including confusion, memory loss and disturbance of attention
- changes in mood, including mood swings
- changes in speech, including difficulty speaking, such as slurred or slow speech
- swelling of the skin or limbs
- problems with your eyes such as difficulty seeing out of one or both eyes, double vision or perceived flashes of light
- feeling of pain, numbness, burning, or pins and needles in your arms or legs
- difficulty walking or performing usual activities such as writing
- diarrhoea
- constipation
- pain in your joints
- tiredness
- weight gain
The above list includes serious side effects that may require medical attention.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people. Some of these (for example, changes in cholesterol and/or triglyceride levels) can only be found when your doctor does tests from time to time to check your progress.
After using LORVIQUA
Storage
Keep your tablets in the pack/ bottle until it is time to take them. If you take the tablets out of the pack/bottle they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store LORVIQUA or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
LORVIQUA 25 mg tablets are round tan coloured film-coated tablets debossed with "Pfizer" on one side and "25" and "LLN" on the other side.
Each blister pack contains 90 or 120 tablets and each bottle contains 30 tablets.
LORVIQUA 100 mg tablets are oval lavender coloured film-coated tablets debossed with "Pfizer" on one side and "LLN 100" on the other side.
Each bottle or blister pack contains 30 tablets.
Ingredients
LORVIQUA tablets contain 25 mg or 100 mg of lorlatinib as the active ingredient. The tablets also contain:
- microcrystalline cellulose
- calcium hydrogen phosphate
- sodium starch glycollate
- magnesium stearate
- hypromellose (E464)
- lactose monohydrate
- macrogol 3350
- triacetin
- titanium dioxide (E171)
- iron oxide black (E172)
- iron oxide red (E172)
Supplier
LORVIQUA is supplied in Australia by:
Pfizer Australia Pty Ltd
Sydney NSW
Toll Free Number: 1800 675 229
www.pfizer.com.au
® = Registered Trademark
This leaflet was prepared in July 2020.
Australian Registration Numbers
LORVIQUA 25 mg:
AUST R 310778 (blister pack)
AUST R 310781 (bottle)
LORVIQUA 100 mg:
AUST R 310780 (blister pack)
AUST R 310779 (bottle)
Published by MIMS September 2020

 A total of 296 patients were randomised to lorlatinib (n=149) or crizotinib (n=147). The demographic characteristics of the overall study population were: median age 59 years (range: 26 to 90 years), age ≥ 65 years (35%), 59% female, 49% White, 44% Asian, and 0.3% Black. The majority of patients had adenocarcinoma (95%) and never smoked (59%). CNS metastases as determined by BICR neuroradiologists were present in 26% (n=78) of patients: of these, 30 patients had measurable CNS lesions.
A total of 296 patients were randomised to lorlatinib (n=149) or crizotinib (n=147). The demographic characteristics of the overall study population were: median age 59 years (range: 26 to 90 years), age ≥ 65 years (35%), 59% female, 49% White, 44% Asian, and 0.3% Black. The majority of patients had adenocarcinoma (95%) and never smoked (59%). CNS metastases as determined by BICR neuroradiologists were present in 26% (n=78) of patients: of these, 30 patients had measurable CNS lesions. The results of prespecified exploratory analyses of intracranial response rate in 30 patients with measurable CNS lesions at baseline as assessed by BICR are summarised in Table 4. Of these, no patients received prior brain radiation.
The results of prespecified exploratory analyses of intracranial response rate in 30 patients with measurable CNS lesions at baseline as assessed by BICR are summarised in Table 4. Of these, no patients received prior brain radiation.

 Among the 93 patients with a confirmed objective response by ICR, the median TTR was 1.4 months (range: 1.1 to 11.0 months). Among the 70 patients with a confirmed objective tumour response by ICR, the median intracranial TTR was 1.4 months (range: 1.1 to 6.2 months).
Among the 93 patients with a confirmed objective response by ICR, the median TTR was 1.4 months (range: 1.1 to 11.0 months). Among the 70 patients with a confirmed objective tumour response by ICR, the median intracranial TTR was 1.4 months (range: 1.1 to 6.2 months).
