What is in this leaflet
This leaflet answers some common questions about Lucentis.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up-to-date information on the medicine. You can also download the most up-to-date leaflet from www.novartis.com.au. Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given Lucentis against the benefits they expect it will have for you.
If you have any concerns about being given this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
What Lucentis is used for
Lucentis is used to treat adults with damage to the retina (light-sensitive layer at the back of the eye) caused by growth of leaky, abnormal blood vessels (choroidal neovascularization, CNV) in diseases that may cause decreased vision such as:
- Wet age related macular degeneration (wet AMD)
- Diabetic macular oedema (DME), or oedema due to retinal vein occlusion (RVO) where fluid accumulates into the back of the eye.
- Proliferative diabetic retinopathy (PDR)
- CNV secondary to pathologic myopia (PM)
- CNV due to other causes, such as angioid streaks, post-inflammatory retinochoroidopathy, central serous chorioretinopathy, and inflammatory CNV.
Lucentis contains the active substance ranibizumab, which is part of an antibody. Antibodies are proteins which specifically recognise and bind to other unique proteins in the body. Ranibizumab binds selectively to a protein called human vascular endothelial growth factor A (VEGF-A), which is present in the retina. It inhibits both the growth and leakage of new blood vessels in the eye, abnormal processes that contribute to several eye diseases that may cause decreased vision.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
It is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children or adolescents.
Before you are given Lucentis
When you must not be given it
You must not be given Lucentis if you have an allergy to:
- any medicine containing ranibizumab
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
You must not be given this medicine if you have or suspect you may have an infection in or around your eye or if you have any pain or redness in your eye.
You must not be given this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering.
If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should be given this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you have allergies to any other medicines, foods, dyes or preservatives.
Tell your doctor if you are pregnant or plan to become pregnant. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you have the potential to become pregnant. It is recommended that you use effective contraception during Lucentis treatment and for at least three months after the last injection of Lucentis.
Tell your doctor if you are breast-feeding or planning to breast-feed. Lucentis is not recommended during breast-feeding as it is not known whether Lucentis passes into breast milk.
Tell your doctor if you have ever had a stroke or have experienced short-lasting signs of a stroke (weakness or paralysis of limbs or face, difficulty speaking or understanding). This information will be taken into account to evaluate if Lucentis is the appropriate treatment for you.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Your doctor and pharmacist have more information on medicines to be careful with or avoid when you are given this medicine.
How Lucentis is given
Lucentis is given by your ophthalmologist (eye doctor) as an injection into your eye under a local anaesthetic.
Follow the directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor for help.
It is important to see your doctor regularly when on treatment with Lucentis.
How much is given
The usual dose is 0.05 mL (equivalent to 0.5mg). The time between two doses injected into the same eye should not be shorter than one month.
The treatment is started with one injection of Lucentis per month. Your doctor will check the condition of your eye. Depending on how you respond to the treatment, your doctor will decide whether and when you need to receive the next injection of Lucentis.
How it is given
Lucentis is given as a single injection into your eye. Before the injection you will be given an eye drop to numb the eye. Before the injection you will also be treated with an eye drop that can kill germs on the eye and on the skin around the eye.
After the Lucentis injection, your doctor may perform some additional tests to make sure there are no complications. Eye injections like those with Lucentis can increase eye pressure. This is something you would not notice.
When it is given
Your doctor will decide when you will be given Lucentis.
How long to continue treatment
Continue treatment with this medicine for as long as your doctor tells you.
If you forget a treatment
If you miss a Lucentis treatment, you need to contact your doctor to arrange another appointment as soon as possible.
If you stop Lucentis treatment, your eye disease may get worse.
If you are given too much (overdose)
If you are given more Lucentis than you need, your doctor will check the pressure in your eye and may need to treat it if it is increased.
While you are being given Lucentis
Things you must do
If you experience any problems during the treatment, tell your doctor.
Tell your doctor immediately if you develop signs of inflammation and/or infection of the eye such as redness of the eye, eye pain, light sensitivity and/or vision changes, seeing flashes of light with floaters (seeing cobwebs), progressing to a loss of sight or blurred vision. A serious eye infection or eye disorder can sometimes develop after an injection into the eye.
If you are treated for visual impairment due to macular oedema in diabetes or in RVO tell your doctor if you think that the effect of the treatment is being lost.
If you become pregnant while being treated with this medicine, tell your doctor immediately.
Things you must not do
Do not drive or operate machinery if your vision is poor, either because of your disease or because of the treatment. This medicine may cause temporary problems with vision in some people. If you are affected, do not drive, operate machinery or do anything else that could be dangerous until your vision is normal.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are being treated with Lucentis.
This medicine helps most people, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- eye irritation, clouding of the lens, a feeling of having something in the eye, dry eye, abnormal sensation in the eye
- eye discomfort, pain or irritation at the site of the injection, increased tear production, redness or itching of the eye, small particles or spots in your vision (floaters)
- sore throat, headache, joint pain, flu-like symptoms, fatigue, general feeling of being unwell, anxiety, cough, nausea, allergic reactions (rash, itching, redness of the skin).
The above list includes side effects of your medicine which are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- bloodshot eye, bleeding in the eye, inflammation or infection of the eyelid margins, visual disturbance, blurred or decreased sharpness of vision, blindness (temporary or otherwise)
- discharge of the eye with itching, redness and swelling (conjunctivitis)
- small marks on the surface of the eye, swelling of a section of the eye (cornea, uvea), swelling or irritation of the eyelid, eyelid pain, sac of pus on the eye.
- symptoms of a urinary tract infection, including burning, stinging, pain or increased urgency to pass water.
The above list includes serious side effects which may require medical attention. Serious side effects are rare.
Tell your doctor immediately if you experience any of the following:
- signs of inflammation or infection of the eye such as redness of the eye, eye pain, sensitivity to light or vision changes
- seeing flashes of light with floaters (seeing spots or cobwebs), progressing to blurred vision or loss of sight.
Go to your nearest emergency room immediately if you experience signs of a stroke, such as weakness or paralysis of limbs or face, difficulty speaking or understanding. If you experience any of these signs, immediate medical care is needed.
The above are serious side effects which might need immediate medical attention.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people. Some of these side effects (e.g. an increase in the pressure inside your eye) can only be found when your doctor does tests to check your progress.
After you are given Lucentis
Storage
It is unlikely you will have to store Lucentis at home.
If you do have to store it:
Vial
- Keep it in a refrigerator (2°C to 8°C). Do not freeze it.
- Keep the vial in the outer carton in order to protect it from light.
- Prior to usage, the unopened vial may be kept at room temperature (25°C) for up to 24 hours.
Pre-filled syringe
- Keep it in a refrigerator (2°C to 8°C). Do not freeze it.
- Keep the pre-filled syringe in its sealed tray in the carton in order to protect it from light.
- Prior to usage, the unopened tray may be kept at room temperature (25°C) for up to 24 hours.
Do not store Lucentis or any other medicine in the bathroom or near a sink.
Do not leave it in the car or on a window sill. Heat and dampness can destroy some medicines.
Keep the medicine where children cannot reach it.
Each Lucentis vial and associated needles and syringe are to be used for one injection only and then discarded.
Product description
What it looks like
Vial pack
Lucentis is supplied as packs containing one glass vial containing 0.23mL of solution, one filter needle for withdrawal of the vial contents, one injection needle and one syringe for intravitreal injection.
Lucentis is a solution for injection supplied in a clear, colourless glass vial. The vial contains 0.23mL of a sterile, clear, colourless to pale yellow aqueous solution.
Vial with filter needle*
Lucentis is supplied as packs containing one glass vial of ranibizumab and one filter needle for withdrawal of the vial contents.
Lucentis is a solution for injection supplied in a clear, colourless glass vial. The vial contains 0.23mL of a sterile, clear, colourless to pale yellow aqueous solution.
Vial*
Lucentis is supplied as packs containing one glass vial of ranibizumab.
Lucentis is a solution for injection supplied in a clear, colourless glass vial. The vial contains 0.23 mL of a sterile, clear, colourless to pale yellow aqueous solution.
Pre-filled syringe
Lucentis is supplied as packs containing one sterile pre-filled syringe in a sealed tray.
Lucentis is a solution for injection supplied in a pre-filled syringe. The pre-filled syringe contains 0.165 mL of a sterile, clear, colourless to pale yellow aqueous solution.
*Not all presentations may be marketed.
Ingredients
Lucentis vial contains 2.3mg ranibizumab as the active ingredient.
Lucentis pre-filled syringe contains 0.165 mg ranibizumab as the active ingredient.
The vial and pre-filled syringe also contains:
- trehalose dihydrate
- histidine hydrochloride monohydrate
- histidine
- polysorbate 20
- water for injections
Allergens:
May contain traces of milk and residue of tetracycline (antibiotic).
Sponsor
Lucentis is supplied in Australia by:
NOVARTIS Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1-800-671-203
Web site: www.novartis.com.au
Date of preparation
This leaflet was prepared in October 2020.
Vial: AUST R 148325 (2.3mg/0.23mL)
Pre-filled syringe: AUST R 212387 (1.65mg/0.165mL)
®= Registered trademark
© Copyright 2020.
Internal document code:
luc261020c based on PI luc261020i.
Published by MIMS December 2020
 A meta-analysis of pooled safety data from completed, randomised, double masked global studies showed a higher incidence rate of non-serious, non-ocular wound infection/inflammation in DME patients treated with ranibizumab 0.5 mg (1.85/100 PY; 20 events in 936 patients) compared to sham/laser treatment (0.27/100 PY; 2 events in 58 patients); HR 8.07 (95% CI 1.88, 34.74). The relationship to ranibizumab remains unknown.
A meta-analysis of pooled safety data from completed, randomised, double masked global studies showed a higher incidence rate of non-serious, non-ocular wound infection/inflammation in DME patients treated with ranibizumab 0.5 mg (1.85/100 PY; 20 events in 936 patients) compared to sham/laser treatment (0.27/100 PY; 2 events in 58 patients); HR 8.07 (95% CI 1.88, 34.74). The relationship to ranibizumab remains unknown. Detailed results are shown in Tables 2 and 3.
Detailed results are shown in Tables 2 and 3.
 Patients in the group treated with Lucentis had minimal observable CNV lesion growth, on average. At month 12, the mean change in the total area of the CNV lesion was 0.1 to 0.3 DA for Lucentis versus 2.3 to 2.6 DA for the control arms.
Patients in the group treated with Lucentis had minimal observable CNV lesion growth, on average. At month 12, the mean change in the total area of the CNV lesion was 0.1 to 0.3 DA for Lucentis versus 2.3 to 2.6 DA for the control arms.


 The long-term safety profile of ranibizumab observed in this 24-month extension study is consistent with the known Lucentis safety profile.
The long-term safety profile of ranibizumab observed in this 24-month extension study is consistent with the known Lucentis safety profile.
 Patients treated with ranibizumab experienced a continuous reduction in central retina thickness. At month 12, the mean CRT change from baseline was -194 micrometres for ranibizumab versus -48 micrometres for sham control.
Patients treated with ranibizumab experienced a continuous reduction in central retina thickness. At month 12, the mean CRT change from baseline was -194 micrometres for ranibizumab versus -48 micrometres for sham control. There was no difference in the BCVA or CRT outcomes of patients in RETAIN study who received or did not receive concomitant thiazolidinediones.
There was no difference in the BCVA or CRT outcomes of patients in RETAIN study who received or did not receive concomitant thiazolidinediones. At year 1 in the ranibizumab-treated group in Protocol S, ≥ 2-step improvement in DRSS was consistent in eyes without DME (39.9%) and with baseline DME (48.8%).
At year 1 in the ranibizumab-treated group in Protocol S, ≥ 2-step improvement in DRSS was consistent in eyes without DME (39.9%) and with baseline DME (48.8%).


 In both studies, the improvement of vision was accompanied by a continuous decrease in the macular oedema as measured by central retinal thickness.
In both studies, the improvement of vision was accompanied by a continuous decrease in the macular oedema as measured by central retinal thickness. In BRIGHTER, 0.5 mg ranibizumab with adjunctive laser therapy demonstrated non-inferiority to ranibizumab monotherapy from baseline to month 24 as assessed by the mean average change in BCVA. There was no difference between the two groups in the number of ranibizumab injections administered over this period.
In BRIGHTER, 0.5 mg ranibizumab with adjunctive laser therapy demonstrated non-inferiority to ranibizumab monotherapy from baseline to month 24 as assessed by the mean average change in BCVA. There was no difference between the two groups in the number of ranibizumab injections administered over this period.
 When comparing ranibizumab versus sham control at month 2, a statistically significant treatment effect for patients in ranibizumab arm was observed.
When comparing ranibizumab versus sham control at month 2, a statistically significant treatment effect for patients in ranibizumab arm was observed. The improvement of vision was accompanied by a reduction in central subfield thickness over the 12-month period.
The improvement of vision was accompanied by a reduction in central subfield thickness over the 12-month period.
 The improvement of vision was accompanied by a reduction in central retinal thickness.
The improvement of vision was accompanied by a reduction in central retinal thickness. During the 24 weeks of the study, fewer patients in the ranibizumab 0.2 mg and 0.1 mg group switched to another treatment modality due to lack of response compared with the laser group (14.9% and 16.9% vs. 24.3%). Unfavourable structural outcomes were less frequently reported for ranibizumab 0.2 mg (1 patient, 1.4%) and 0.1 mg (5 patients, 6.7%) compared with laser therapy (7 patients, 10.1%). The recurrence of ROP (as measured by the need for any post-baseline intervention at or before 24 weeks) was 31.1%, 31.2% and 18.9% in the ranibizumab 0.2 mg, ranibizumab 0.1 mg and laser groups, respectively. At week 40, these percentages were 31.1%, 33.8% and 20.4% in the ranibizumab 0.2 mg, ranibizumab 0.1 mg and laser groups, respectively.
During the 24 weeks of the study, fewer patients in the ranibizumab 0.2 mg and 0.1 mg group switched to another treatment modality due to lack of response compared with the laser group (14.9% and 16.9% vs. 24.3%). Unfavourable structural outcomes were less frequently reported for ranibizumab 0.2 mg (1 patient, 1.4%) and 0.1 mg (5 patients, 6.7%) compared with laser therapy (7 patients, 10.1%). The recurrence of ROP (as measured by the need for any post-baseline intervention at or before 24 weeks) was 31.1%, 31.2% and 18.9% in the ranibizumab 0.2 mg, ranibizumab 0.1 mg and laser groups, respectively. At week 40, these percentages were 31.1%, 33.8% and 20.4% in the ranibizumab 0.2 mg, ranibizumab 0.1 mg and laser groups, respectively.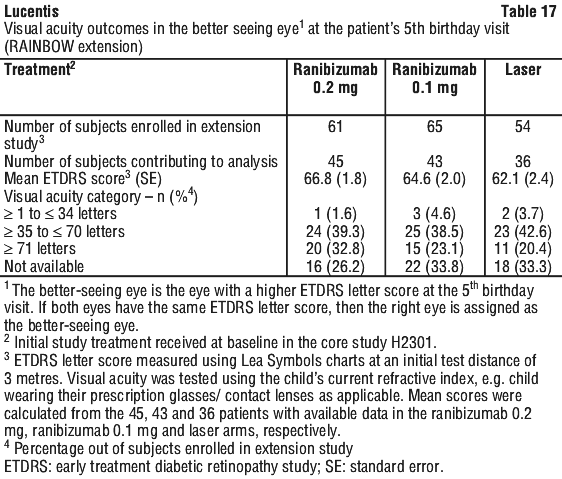

 Chemical name: Immunoglobulin G1, anti-(human vascular endothelial growth factor) Fab fragment (human-mouse monoclonal rhuFab V2 γ1-chain), disulfide with human-mouse monoclonal rhuFab V2 κ-chain.
Chemical name: Immunoglobulin G1, anti-(human vascular endothelial growth factor) Fab fragment (human-mouse monoclonal rhuFab V2 γ1-chain), disulfide with human-mouse monoclonal rhuFab V2 κ-chain.