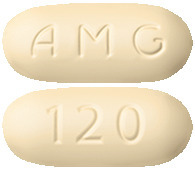1 Name of Medicine
Sotorasib.
2 Qualitative and Quantitative Composition
Active substance.
Lumakras tablet.
Each film coated tablet contains sotorasib 120 mg.
Excipients.
Excipient with known effect.
Contains sugars. Each Lumakras tablet contains lactose (see Section 4.4 Special Warnings and Precautions for Use).
For the list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Lumakras 120 mg tablet.
Yellow, immediate release, film coated tablet, oblong-shaped (approximately 7 mm x 16 mm), debossed with "AMG" on one side and "120" on the reverse side.4.1 Therapeutic Indications
Lumakras has provisional approval in Australia for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC) who have received at least one prior systemic therapy for advanced disease.
The decision to approve this indication has been made on the basis of the objective response rate (ORR) and the duration of response (DOR). Continued approval of this indication depends on the verification and description of benefit in confirmatory trials.
4.2 Dose and Method of Administration
Confirm the presence of a KRAS G12C mutation using a validated test prior to initiation of Lumakras.
Dose.
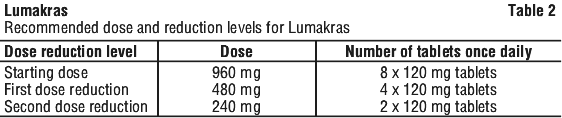
The recommended dose of Lumakras is 960 mg (as eight 120 mg tablets) orally once daily until disease progression or unacceptable toxicity (see Table 2).
Take Lumakras at the same time each day with or without food. Swallow tablets whole. Do not chew, crush, or split tablets.
If a dose of Lumakras is missed, do not take the dose if 6 hours or more have passed from the scheduled time of dosing. Resume treatment as prescribed the next day.
If vomiting occurs after taking Lumakras, do not take an additional dose. Resume treatment as prescribed the next day.
Administration to patients who have difficulty swallowing solids.
Disperse tablets in 120 mL of room-temperature tap water without crushing. Do not use other liquids. Stir until tablets are dispersed into small pieces (the tablets will not completely dissolve) and drink immediately or within two hours of preparation. The appearance of the mixture may range from pale yellow to bright yellow. Swallow the tablet dispersion. Do not chew pieces of the tablet. Rinse the container with an additional 120 mL of water and drink immediately. If the mixture is not consumed immediately, stir the mixture again to ensure that tablets are dispersed.
Dose modifications.
Lumakras dose modifications for adverse reactions are provided in Table 1. Lumakras dose reduction levels are summarised in Table 2. If adverse reactions occur, a maximum of two dose reductions are permitted.
Discontinue Lumakras if patients are unable to tolerate the minimum dose of 240 mg once daily.

Special populations.
Hepatic impairment.
No dosage modification is recommended in patients with mild to moderate hepatic impairment (Child-Pugh A or B).
The effect of severe hepatic impairment (Child-Pugh C) on the safety of Lumakras is unknown. Monitor for sotorasib adverse reactions in patients with hepatic impairment more frequently since these patients may be at increased risk for adverse reactions including hepatotoxicity.
Renal impairment.
Based on population pharmacokinetic analysis, no dose adjustment is recommended for patients with mild renal impairment (creatinine clearance ≥ 60 mL/min) (see Section 5.2 Pharmacokinetic Properties). Lumakras has not been studied in patients with moderate or severe (creatinine clearance: < 60 mL/min) renal impairment (see Section 5.2 Pharmacokinetic Properties).
Paediatric use.
The safety and efficacy of Lumakras in paediatric patients have not been established.
Use in the elderly.
In clinical studies, no overall differences in Lumakras safety or efficacy were observed between geriatric patients (≥ 65 years old) and younger patients. No dose adjustment is required for geriatric patients (see Section 5.2 Pharmacokinetic Properties).
Coadministration of Lumakras with acid-reducing agents.
Avoid coadministration of proton pump inhibitors (PPIs) and H2-receptor antagonists with Lumakras. If treatment with an acid-reducing agent cannot be avoided, take Lumakras 4 hours before or 10 hours after administration of a local antacid (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions, Acid-reducing agents; Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
Lumakras is contraindicated in patients with hypersensitivity to sotorasib or any of the excipients (see Section 6.1 List of Excipients).
4.4 Special Warnings and Precautions for Use
Lactose.
Lumakras tablets contain lactose monohydrate 114 mg (see Section 2 Qualitative and Quantitative Composition; Section 6.1 List of Excipients). Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Hepatotoxicity.
Lumakras can cause hepatotoxicity and increased alanine aminotransferase (ALT) or increased aspartate aminotransferase (AST) which may lead to drug induced liver injury and hepatitis.
In the pooled safety population of patients with NSCLC who received single agent Lumakras 960 mg [see Section 4.8 Adverse Effects (Undesirable Effects)], hepatotoxicity occurred in 27% of patients, of which 16% were Grade ≥ 3. Among patients with hepatotoxicity who required dosage modifications, 64% required treatment with corticosteroids.
In the pooled safety population of patients with NSCLC who received single agent Lumakras 960 mg, 17% of patients who received Lumakras had increased ALT/increased AST; of which 9% were Grade ≥ 3. The median time to first onset of increased ALT/AST was 6.3 weeks (range: 0.4 to 42). Increased ALT/AST leading to dose interruption or reduction occurred in 9% of patients. Lumakras was discontinued due to increased ALT/AST in 2.7% of patients. Drug-induced liver injury occurred in 1.6% (all grades) including 1.3% (Grade ≥ 3).
In this pooled safety population of patients with NSCLC who received single agent Lumakras 960 mg, a total of 40% patients with recent (≤ 3 months) immunotherapy prior to starting Lumakras had an event of hepatotoxicity. An event of hepatotoxicity was observed in 18% of patients who started Lumakras more than 3 months after last dose of immunotherapy and in 17% of those who never received immunotherapy. Regardless of time from prior immunotherapy, 94% of hepatotoxicity events improved or resolved with dosage modification of Lumakras, with or without corticosteroid treatment.
Monitor liver function tests (ALT, AST, alkaline phosphatase and total bilirubin) prior to the start of Lumakras, every 3 weeks for the first 3 months of treatment, then once a month or as clinically indicated, with more frequent testing in patients who develop transaminase and/or bilirubin elevations. Withhold, dose reduce or permanently discontinue Lumakras based on severity of adverse reaction [see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects)].
Consider administering systemic corticosteroids for the management of hepatotoxicity.
Interstitial lung disease (ILD)/pneumonitis.
In the pooled safety population of patients with NSCLC who received single agent Lumakras 960 mg [see Section 4.8 Adverse Effects (Undesirable Effects)], ILD/pneumonitis occurred in 2.2% of patients, of which 1.1% were Grade ≥ 3, and 1 case was fatal. The median time to first onset for ILD/pneumonitis was 8.6 weeks (range: 2.1 to 36.7 weeks). Lumakras was permanently discontinued due to ILD/pneumonitis in 1.3% of patients.
Recent (≤ 3 months) immunotherapy prior to starting Lumakras may be considered a risk factor for ILD/pneumonitis. Monitor patients for new or worsening pulmonary symptoms indicative of ILD/ pneumonitis (e.g. dyspnoea, cough, fever). Immediately withhold Lumakras in patients with suspected ILD/pneumonitis and permanently discontinue Lumakras if no other causes of ILD/pneumonitis are identified (see Section 4.2 Dose and Method of Administration).
Effects on laboratory tests.
Sotorasib has been associated with transient elevations of ALT and AST.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of other medicines on Lumakras.
Acid-reducing agents.
The solubility of sotorasib is pH-dependent. Coadministration of Lumakras with gastric acid-reducing agents decreased sotorasib concentrations, which may reduce the efficacy of sotorasib. Avoid coadministration of Lumakras with proton pump inhibitors (PPIs), H2-receptor antagonists, and locally acting antacids. If coadministration with an acid-reducing agent cannot be avoided, administer Lumakras 4 hours before or 10 hours after administration of a locally acting antacid (see Section 5.2 Pharmacokinetic Properties).
Strong CYP3A inducers.
Coadministration of Lumakras with a strong CYP3A inducer led to a decrease in sotorasib concentrations. Coadministration of strong CYP3A4 inducers (including rifampin, carbamazepine, enzalutamide, phenobarbital, and St John's wort) with Lumakras is not recommended because the impact on efficacy is unknown (see Section 5.2 Pharmacokinetic Properties).
Effect of Lumakras on other medicines.
CYP3A4 substrates.
Lumakras is a moderate CYP3A4 inducer. Coadministration of Lumakras with CYP3A4 substrates (such as alfentanil, fentanyl, cyclosporin, sirolimus, everolimus, tacrolimus, simvastatin, atorvastatin, midazolam, amiodarone, rivaroxaban and apixaban) could lead to a decrease in their plasma concentrations, which may reduce the efficacy of these substrates (see Section 5.2 Pharmacokinetic Properties). Avoid coadministration of Lumakras with CYP3A4 substrates with narrow therapeutic indices. If coadministration cannot be avoided, adjust the CYP3A4 substrate dosage in accordance with approved product labelling.
BCRP substrates.
Lumakras is a BCRP inhibitor. Coadministration of Lumakras with a BCRP substrate led to an increase in the plasma concentrations of the BCRP substrate, which may increase the effects of these substrates (see Section 5.2 Pharmacokinetic Properties). When co-administered with Lumakras, monitor for adverse reactions of the BCRP substrate and decrease the BCRP substrate dosage in accordance with its product labelling.
P-glycoprotein (P-gp) substrates.
Coadministration of Lumakras with a P-gp substrate (digoxin) increased digoxin plasma concentrations [see Section 5.2 Pharmacokinetic Properties, Effect of sotorasib on other medicines], which may increase the adverse reactions of digoxin. Avoid coadministration of Lumakras with P-gp substrates, for which minimal concentration changes may lead to serious toxicities. If coadministration cannot be avoided, decrease the P-gp substrate dosage in accordance with its Product Information.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no clinical studies to evaluate the effect of Lumakras on fertility. Fertility/early embryonic development studies were not conducted with sotorasib. There were no adverse effects on female or male reproductive organs in general toxicology studies conducted in dogs and rats.
(Category B3)
There are no clinical studies with Lumakras use in pregnant women. In rat and rabbit embryo-fetal development studies, oral sotorasib was not teratogenic. Inform the patient of the potential hazards to the fetus if Lumakras is used during pregnancy, or if the patient becomes pregnant while taking Lumakras.
Animal data.
In the rat, there were no effects on embryo-fetal development up to 540 mg/kg/day the highest dose tested [approximately 2 times higher than the exposure at the maximum recommended human dose (MRHD) of sotorasib 960 mg, based on area under the curve, AUC].
In the rabbit, lower fetal body weights and a reduction in the number of ossified metacarpals in fetuses were observed only at the highest dose level tested (100 mg/kg, 2.3 times higher than the exposure at the MRHD of 960 mg based on AUC), which was associated with maternal effects such as decreased body weight gain and decreased food consumption during the dosing phase. Reduced ossification, as evidence of growth retardation associated with reduced fetal body weight, was interpreted as a non-specific effect in the presence of significant maternal toxicity.
There are no clinical studies on the presence of Lumakras or its metabolites in human milk, the effects on the breastfed child, or on milk production. Because of the potential risk for Lumakras to cause adverse effects in breastfed children, advise women not to breastfeed during treatment with Lumakras and for 1 week after the final dose.4.7 Effects on Ability to Drive and Use Machines
Lumakras has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Summary of safety profile.
NSCLC (pooled analysis).
The pooled safety population described in the Section 4.4 Special Warnings and Precautions for Use reflect exposure to Lumakras as a single agent at 960 mg orally once daily until disease progression or unacceptable toxicity in 549 patients with NSCLC with KRAS G12C mutation in the following trials: CodeBreaK 200, CodeBreaK 100, CodeBreaK 101 and CodeBreaK 105. The median duration of exposure to Lumakras was 4.8 months (range: 0 to 41 months).
Locally advanced or metastatic NSCLC (CodeBreak 100).
The safety of Lumakras was evaluated in 214 patients with KRAS G12C-mutated locally advanced or metastatic NSCLC who received sotorasib 960 mg orally once daily as monotherapy in CodeBreak 100. The median duration of exposure to Lumakras was 5.5 months (range: 0.2 to 21 months).
Serious adverse reactions occurred in 50% of patients treated with Lumakras. Serious adverse reactions in ≥ 2% of patients were pneumonia (8%), hepatotoxicity (3.4%), and diarrhoea (2%). Fatal adverse reactions occurred in 3.4% of patients who received Lumakras due to respiratory failure (0.8%), pneumonitis (0.4%), cardiac arrest (0.4%), cardiac failure (0.4%), gastric ulcer (0.4%), and pneumonia (0.4%).
Permanent discontinuation of Lumakras due to an adverse reaction occurred in 9% of patients. Adverse reactions resulting in permanent discontinuation of Lumakras in ≥ 2% of patients included hepatotoxicity (4.9%).
Dosage interruptions of Lumakras due to an adverse reaction occurred in 34% of patients. Adverse reactions which required dosage interruption in ≥ 2% of patients were hepatotoxicity (11%), diarrhoea (8%), musculoskeletal pain (3.9%), nausea (2.9%), and pneumonia (2.5%).
Dose reductions of Lumakras due to an adverse reaction occurred in 5% of patients. Adverse reactions which required dose reductions in ≥ 2% of patients included increased ALT (2.9%) and increased AST (2.5%).
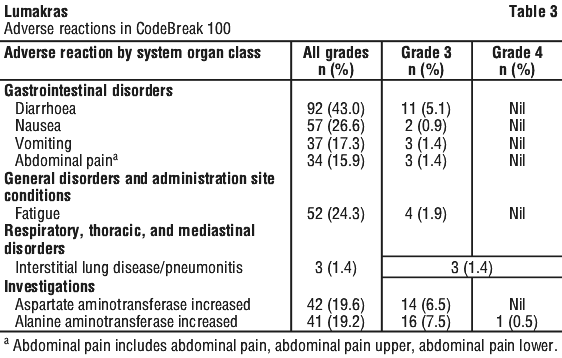
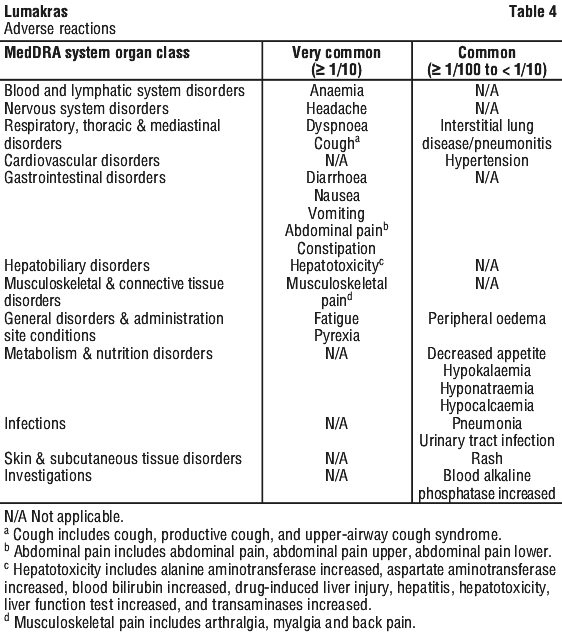
Adverse drug reactions reported by system organ class in Lumakras clinical studies are displayed in Table 3.
Tabulated list of adverse reactions.
Adverse reactions reported in Lumakras clinical studies are displayed in Table 4. Frequency is provided by MedDRA category: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 and < 1/1,000), very rare (< 10,000). Within each system organ class, adverse reactions are presented in order of decreasing seriousness.
Reporting of suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at https://www.tga.gov.au/reporting-problems.4.9 Overdose
Symptoms and signs.
There is no clinical experience of overdose with Lumakras.
Treatment.
In the event of a Lumakras overdose, the patient should be treated symptomatically, and supportive measures instituted as required. For advice on the management of overdose contact the Poisons Information Centre on 131126.5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antineoplastic agent.
Anatomical Therapeutic Chemical (ATC) code: L01XX73.
Mechanism of action.
Sotorasib is a KRASG12C inhibitor, which covalently and irreversibly binds to the unique cysteine of KRASG12C. Inactivation of KRASG12C by sotorasib blocks tumour cell signalling and survival, inhibits cell growth, and promotes apoptosis selectively in tumours harbouring KRASG12C, an oncogenic driver of tumourigenesis across multiple cancer types. The potency and selectivity of sotorasib is enhanced through the unique binding to both the P2 pocket and the His95 surface groove, locking the protein in an inactive state that prevents downstream signalling without affecting wild-type KRAS.
Sotorasib demonstrated in vitro and in vivo inhibition of KRASG12C with minimal detectable off-target activity against other cellular proteins and processes. Sotorasib impaired oncogenic signalling and tumour cell survival at clinically relevant exposures in preclinical models expressing KRASG12C. Sotorasib also enhanced antigen presentation and inflammatory cytokine production only in tumour cells with KRASG12C. Sotorasib induced anti-tumour inflammatory responses and immunity, driving tumour regressions in immunocompetent mice implanted with KRASG12C-expressing tumours.
Cardiac electrophysiology.
The effect of Lumakras on the QT interval was assessed in 156 patients administered Lumakras 960 mg once daily in clinical studies. Lumakras did not prolong the QT interval to any clinically relevant extent. At peak concentrations, the mean change from baseline was less than 5 milliseconds (ms). No patients had a large mean increase in QTc (> 20 ms) in the studies.
Clinical trials in NSCLC.
CodeBreaK 100.
The efficacy of Lumakras was demonstrated in a single-arm, open-label, multicentre trial (CodeBreaK 100) that enrolled patients with locally advanced or metastatic KRAS G12C-mutated NSCLC who had disease progression on or after receiving prior therapy. Key eligibility criteria included progression on an immune checkpoint inhibitor and/or platinum-based chemotherapy, an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1, and at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumours (RECIST v1.1).
All patients were required to have KRAS G12C-mutated NSCLC prospectively identified in tumour samples by a validated test performed in a central laboratory. From the patients with KRAS G12C mutations confirmed in tumour tissue, plasma samples from 112 patients were tested retrospectively using a separate validated test. 78 patients (70%) had KRAS G12C mutation identified in plasma specimen, and 31 patients (28%) did not have KRAS G12C mutation identified in plasma specimen.
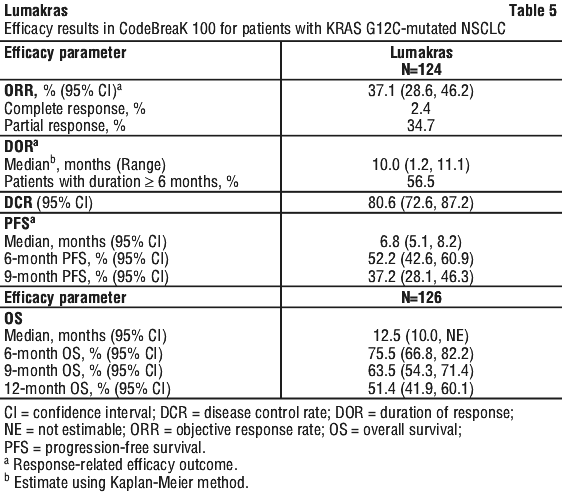
A total of 126 patients were enrolled and treated with Lumakras 960 mg once daily until disease progression or unacceptable toxicity; 124 patients had at least one measurable lesion at baseline as assessed by Blinded Independent Central Review (BICR) according to RECIST v1.1 and were included in the analysis for response-related efficacy outcomes. The median duration of treatment was 5.5 months (range 0 to 15) with 48% of patients treated for ≥ 6 months and 33% of patients treated for ≥ 9 months. The major efficacy outcome measure was objective response rate (ORR) and duration of response (DOR) as evaluated by BICR according to RECIST v1.1. Additional efficacy outcome measures included disease control rate (DCR), time to response (TTR), progression-free survival (PFS), and overall survival (OS).
The baseline demographic and disease characteristics of the study population were: median age 64 years (range 37 to 80) with 47% ≥ 65 years and 8% ≥ 75 years; 50% Female; 82% White, 15% Asian, 2% Black; 70% ECOG PS 1; 96% had stage IV disease; 99% with non squamous histology; 81% former smokers, 12% current smokers, 5% never smokers. All patients received at least 1 prior line of systemic therapy for metastatic NSCLC; 43% received only 1 prior line of therapy, 35% received 2 prior lines of therapy, 22% received 3 prior lines of therapy; 91% received prior anti-PD-1/PD-L1 immunotherapy, 90% received prior platinum-based chemotherapy, 81% received both platinum-based chemotherapy and anti-PD-1/PD-L1. The sites of known extra-thoracic metastasis included 48% bone, 21% brain, and 21% liver.
Efficacy results are summarised in Table 5. The ORR was 37% (95% CI: 29, 47). The patients with objective responses had DOR ranging from 1.2 to 11.1 months, and 43% were still on therapy with ongoing response after a median duration of follow-up of 9.6 months. The median TTR was 1.4 months (range 1.2 to 10.1), with 70% of responses occurring within the first 7 weeks. Consistent efficacy results were seen in patients with KRAS G12C mutation identified in either tissue or plasma specimens.
5.2 Pharmacokinetic Properties
The pharmacokinetics of sotorasib have been characterised in patients with KRAS G12C-mutated solid tumours, including NSCLC, and healthy subjects.
In a dose comparison sub-study in patients receiving sotorasib 960 mg or 240 mg once daily dose, after 8 daily doses, geometric mean Cmax and AUC0-24 for the 240 mg dose were both 22% lower than for the 960 mg dose.
Absorption.
Following an oral, single-dose administration, sotorasib was absorbed with median time to achieve peak concentration of 1 hour.
Effect of food.
Following administration of sotorasib with a high-fat, high-calorie meal, there was no effect on Cmax and AUC increased by 38% compared to administration under fasted conditions. Sotorasib can be administered with or without food.
Distribution.
The mean volume of distribution at steady state of sotorasib was 211 L. In vitro, plasma protein binding of sotorasib was 89%.
Metabolism.
The main metabolic pathways of sotorasib were non-enzymatic conjugative and oxidative metabolism by CYP3As.
Excretion.
At sotorasib 960 mg once daily, the steady state apparent clearance is 26.2 L/hr. The mean half-life is 5 hours. Steady state was reached within 22 days and remained stable. No accumulation with multiple dosing was observed. Sotorasib is primarily eliminated in faeces, with approximately 74% of the dose recovered in faeces and 6% (1% unchanged) recovered in urine.
Special populations.
No clinically meaningful differences in the pharmacokinetics of sotorasib were observed based on age, sex, race, body weight, line of therapy, ECOG PS, mild renal impairment (creatinine clearance: ≥ 60 mL/min), or mild hepatic impairment (AST or ALT < 2.5 x ULN or total bilirubin < 1.5 x ULN). The effect of moderate to severe renal or hepatic impairment on sotorasib pharmacokinetics has not been studied.
Hepatic impairment.
The mean AUC of sotorasib decreased by 25% in subjects with moderate hepatic impairment (Child-Pugh B) and increased by 4% in subjects with severe hepatic impairment (Child-Pugh C) compared to subjects with normal hepatic function following a single dose of 960 mg Lumakras.
Drug interaction studies.
Effect of other medicines on sotorasib. Acid-reducing agents: Coadministration of repeat doses of omeprazole (PPI) with a single dose of Lumakras decreased sotorasib Cmax by 65% and AUC by 57% under fed conditions, and decreased sotorasib Cmax by 57% and AUC by 42% under fasted conditions. Coadministration of a single dose of famotidine (H2-receptor antagonist) given 10 hours prior to and 2 hours after a single dose of Lumakras under fed conditions decreased sotorasib Cmax by 35% and AUC by 38% (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Strong CYP3A4 inducers.
Coadministration of Lumakras with multiple doses of rifampin (a strong CYP3A4 inducer) decreased sotorasib Cmax by 35% and AUC by 51% (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Strong CYP3A4 inhibitors and transporter systems.
No clinically meaningful effect on the exposure of sotorasib was observed following coadministration of Lumakras with itraconazole (a strong CYP3A4 inhibitor and P-glycoprotein [P-gp] inhibitor), single dose of rifampin [an organic-anion-transporting polypeptides (OATP) OATP1B1/1B3 inhibitor], or metformin (a multidrug and toxin extrusion (MATE) MATE1/MATE2-K substrate).
Effect of sotorasib on other medicines. CYP3A4 substrates.
Coadministration of Lumakras with midazolam (a sensitive CYP3A4 substrate) decreased midazolam Cmax by 48% and AUC by 53% (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
BCRP substrates.
Coadministration of Lumakras with rosuvastatin (a BCRP substrate) increased rosuvastatin Cmax by 70% and AUC by 34% (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
P-gp substrates.
Coadministration of Lumakras with digoxin (a P-gp substrate) increased digoxin Cmax by 91% and AUC by 21%.
Transporter systems.
No clinically meaningful effect on the exposure of metformin (a MATE1/MATE2-K substrate) or digoxin (a sensitive P-gp substrate) were observed following coadministration of Lumakras.
In vitro studies. Cytochrome P450 (CYP) enzymes.
Sotorasib may induce CYP2C8, CYP2C9, CYP2C19 and CYP2B6. Sotorasib does not inhibit CYP1A2, CYP2B6, CYP2C9, or CYP2C19 at clinically relevant concentrations. Sotorasib inhibited CYP2D6 with Ki=18.2 microM.
Transporter systems.
Sotorasib may have the potential to inhibit breast cancer resistance protein (BCRP), OATP1B1, OATP1B3, OCT1, and OAT3; the clinical relevance of these findings is unknown.
5.3 Preclinical Safety Data
Reproductive toxicity and fertility.
Fertility/early embryonic development studies were not conducted with sotorasib. There were no adverse effects on male or female reproductive organs in general toxicology studies conducted in dogs and rats.
Genotoxicity.
Sotorasib was not mutagenic in a bacterial mutagenicity (Ames) assay. Sotorasib was not genotoxic in the in vivo rat micronucleus and comet assays.
Carcinogenicity.
Carcinogenicity studies have not been performed with sotorasib.6 Pharmaceutical Particulars
6.1 List of Excipients
The core tablets contain microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate (vegetable source). The film coating contains polyvinyl alcohol, titanium dioxide, polyethylene glycol, purified talc, and iron oxide yellow.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
Information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG).
The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Lumakras tablets are supplied in either PVC/PVDC/Aluminium or PVC/Aclar/ Aluminium blister packs. Each blister strip contains 8 film coated tablets. Each pack contains 240 film coated tablets (30 blister strips).
6.6 Special Precautions for Disposal
Any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
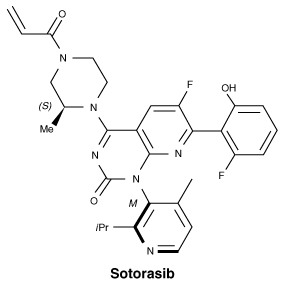 Sotorasib contains one asymmetric centre and one chiral axis in the indicated (S)- and (M)- configuration.
Sotorasib contains one asymmetric centre and one chiral axis in the indicated (S)- and (M)- configuration.
Chemical name (IUPAC): 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4- [(2S)-2- methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one.
Molecular formula: C30H30F2N6O3.
Molecular weight: 560.6 Daltons.
CAS number.
2296729-00-3.
Sotorasib is a white, off-white, or yellow to light brown crystalline powder with low hygroscopicity. The melting point is approximately 289°C.
Sotorasib is almost insoluble in water. The solubility of sotorasib in aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL. An aqueous 0.06 mg/mL solution has a pH of 5.6. Sotorasib has pKa values of 4.56 and 8.06. The partition coefficient (log D) at pH 7.4 is 2.44.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes