

What is in this leaflet
This leaflet answers some common questions about LYNPARZA tablets. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking LYNPARZA tablets against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What LYNPARZA tablets are used for
LYNPARZA tablets are used to treat
- a certain type of ovarian cancer (BRCA-mutated) that is newly diagnosed. It is used once the cancer has responded to treatment with standard platinum-based chemotherapy. A test is used to determine whether you have BRCA-mutated ovarian cancer.
- ovarian cancer that has recurred and it has responded to previous treatment with platinum-based chemotherapy.
- ovarian cancer in combination with another anti-cancer medicine called bevacizumab. These medicines are used together once the cancer has responded to the first treatment with standard platinum-based chemotherapy.
- a certain type of breast cancer (germline BRCA-mutated, human epidermal growth factor receptor 2-negative (HER2-negative)) which has spread beyond the original tumour. You should have received chemotherapy medicines either before or after your cancer has spread. A test is used to determine if you have germline BRCA mutated breast cancer.
- a certain type of pancreatic cancer (germline BRCA-mutated) which has spread beyond the original tumour. It is used if the cancer has not progressed after treatment with chemotherapy. A test is used to determine whether you have BRCA-mutated pancreatic cancer.
- a certain type of prostate cancer (BRCA-mutated) which has spread beyond the original tumour and no longer responds to medical or surgical treatment that lowers testosterone. You should have already received certain hormonal treatments such as abiraterone acetate or enzalutamide. A test is used to determine whether you have this type of prostate cancer.
LYNPARZA belongs to a group of medicines called PARP (Poly (ADP-Ribose) Polymerase enzymes) inhibitors. PARP inhibitors can destroy cancer cells that are not good at repairing DNA damage. These specific cancer cells can be identified by response to platinum chemotherapy or by looking for faulty DNA repair genes such as BRCA (BReast CAncer) genes.
When LYNPARZA is given in combination with other anti-cancer medicines it is important that you also read the package leaflets of these other medicines. If you have any questions about these medicines, ask your doctor.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
Before you take LYNPARZA tablets
When you must not take it
Do not take LYNPARZA tablets if you have an allergy to:
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take this medicine if you are pregnant or if you (male or female) are planning to have a baby. It may affect your developing baby if you take it during pregnancy. Your doctor will advise women to ensure they are using effective contraception when using the medicine and for at least 1 month after stopping the medicine. Male patients and their partners must use effective contraception when using the medicine for at least 3 months after stopping the medicine.
Do not breast-feed if you are taking this medicine. It is not known if the active ingredient in LYNPARZA tablets passes into breast milk. Breast-feeding mothers are advised not to breast-feed during treatment with LYNPARZA tablets and for one month after receiving the last dose.
Do not give this medicine to children. Safety and effectiveness in children younger than 18 years have not been established.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- problems with your kidneys or liver (renal or hepatic impairment)
- problems with your blood (e.g. anaemia, low white blood cell counts).
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding.
If you are a man, tell your doctor if you are planning to have a baby with your partner. Your doctor can discuss with you the risks and benefits involved. Whether you are male or female, your doctor will advise you to use an effective form of contraception while taking LYNPARZA and for at least 1 month for women and 3 months for men after stopping treatment. Female partners of male patients taking LYNPARZA must also use a suitable method of contraception. Male patients must not donate sperm whilst taking LYNPARZA and for 3 months after stopping treatment.
If you have not told your doctor about any of the above, tell them before you start taking LYNPARZA tablets.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines or foods and LYNPARZA may interfere with each other and reduce effectiveness. These include:
- medicines used to treat fungal infections (antifungals) with active ingredients such as fluconazole, ketoconazole, itraconazole
- medicines used to treat bacterial infections (antibiotics) with active ingredients such as clarithromycin, rifampicin, rifabutin, ciprofloxacin, erythromycin
- medicines used to treat viral infections (especially HIV) with active ingredients such as ritonavir, indinavir, saquinavir, nevirapine, boceprevir, cobicistat, etravirine and efavirenz
- medicines used to treat epilepsy with active ingredients such as phenytoin, carbamazepine
- medicines with active ingredients such as modafinil used to treat a sleep disorder called narcolepsy (where you may experience very sleepy periods at odd times during the day)
- medicines to treat high blood pressure, angina (chest pain), irregular heartbeat or heart failure such as diltiazem, digoxin, furosemide, valsartan and verapamil
- medicines called statins used to treat high cholesterol such as rosuvastatin and atorvastatin
- medicines used to treat diabetes, such as glibenclamide and metformin
- bosentan, a medicine used to treat high blood pressure in the lungs
- medicines used to suppress the immune system, such as ciclosporin, tacrolimus and sirolimus
- fentanyl, a medicine used to treat cancer pain
- methotrexate, a medicine used to treat cancer, rheumatoid arthritis and psoriasis
- quetiapine, a medicine used to treat mental disorders
- colchicine, a medicine used to treat gout
- dabigatran, a medicine to prevent blood clots
- grapefruit, Seville oranges and star fruit
- St John's Wort, used to treat depression.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
If you have not told your doctor about any of these things, tell them before you take any LYNPARZA tablets.
How to take LYNPARZA tablets
Your doctor has prescribed LYNPARZA tablets for you.
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the pack, ask your doctor or pharmacist for help.
How much to take
The usual dose is two 150 mg tablets taken twice each day (a total of 4 tablets each day).
Your doctor may prescribe a different dose if you have problems with your kidneys, or are taking certain medicines that may interact with LYNPARZA tablets or if you experience certain side effects while you are taking LYNPARZA tablets.
Your doctor will tell you how many tablets of LYNPARZA to take and it is important that you take the total recommended daily dose.
How to take it
Swallow the tablets whole with a glass of water. Do not chew, crush, dissolve or divide the tablets as this may affect how quickly the drug gets into your body.
When to take it
Take LYNPARZA tablets at about the same time each morning and evening. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
LYNPARZA tablets can be taken with or without food.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
It is important to keep taking your medicine even if you feel well.
If you forget to take it
If you forget to take LYNPARZA, take your next normal dose at its scheduled time. Do not take a double dose (two doses at the same time) to make up for forgotten tablets.
If you are not sure what to do, ask your doctor.
If you have trouble remembering to take your medicine, ask your doctor or pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much LYNPARZA. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using LYNPARZA tablets
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking LYNPARZA.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine or within 1 month after receiving the last dose of LYNPARZA, tell your doctor immediately.
If you are a man and your partner becomes pregnant while you are taking this medicine or within 3 months of stopping this medicine, tell your doctor immediately. Ensure you are using effective contraception. Even if you stop treatment, you should continue to use effective contraception for at least a further 1 month for female patients and 3 months for male patients.
Male patients must use a condom whilst taking LYNPARZA and for 3 months after receiving the last dose when having sexual intercourse with a female partner, even if they are pregnant. Your female partner must also use a suitable method of contraception. Male patients must not donate sperm whilst taking LYNPARZA and for 3 months after receiving the last dose.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects. Your doctor will test your blood every month for the first year of treatment and periodically thereafter.
Things you must not do
Do not take LYNPARZA tablets to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor. If you stop taking it suddenly, your condition may worsen.
Your doctor may interrupt your treatment or reduce your dose if you are having unwanted side effects.
Things to be careful of
Be careful driving or operating machinery until you know how LYNPARZA affects you. This medicine may cause dizziness and tiredness in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking LYNPARZA tablets.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor straight away if you notice any of the following:
- feeling or being sick (nausea or vomiting)
- dizziness, tiredness or weakness
- indigestion or heartburn
- loss of appetite
- headache
- change in taste of food (if it worries you)
- diarrhoea
- sore mouth
- cough
- pain in the stomach area under the ribs
- shortness of breath and/or a dry cough which can be due to inflammation of the lungs (pneumonitis).
- rash itchy swollen reddened skin
- facial swelling
The above list includes serious side effects that may require medical attention and can be life-threatening, especially if not treated.
If any of the following happen, tell your doctor immediately, or go to Accident and Emergency at your nearest hospital:
- tightness of the chest, wheezing, coughing or difficulty breathing.
- swelling of the face, lips, tongue or other parts of the body.
- severe skin reaction which may include rash, itching, redness, blistering or peeling of the skin.
These are very serious side effects. If you have them, you may have had a serious allergic reaction to LYNPARZA. You may need urgent medical attention or hospitalisation.
Some other serious side effects may only become known through tests. Your doctor will test your blood every month for the first year of treatment and periodically thereafter. The blood tests may show:
- a condition where there is damage to the blood-forming cells in your bone marrow (myelodysplastic syndrome/acute myeloid leukaemia)
- decrease in the number of red blood cells (anaemia) which can be associated with shortness of breath, fatigue, pale skin, or fast heart beat
- decrease in the number of white blood cells which can be associated with fever or infection
- increase in blood creatinine which can mean your kidneys are not working as well
- decrease in the number of platelets, which can result in bruising or bleeding for longer than normal if injured.
These conditions may also be life-threatening, especially if not treated.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Your doctor may prescribe other medicines to control unwanted side effects.
After using LYNPARZA tablets
Storage
Keep your tablets in the blister pack until it is time to take them. If you take the tablets out of the blister they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store LYNPARZA tablets or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
LYNPARZA 100 mg tablet is a yellow to dark yellow, oval, bi-convex, film-coated tablet, marked with 'OP100' on one side and plain on the other.
LYNPARZA 150 mg tablet is a green to green/grey oval, bi-convex, film-coated tablet, marked with 'OP150' on one side and plain on the other.
LYNPARZA tablets are available in blisters containing 56 film-coated tablets, packed in cartons containing 7 blisters of 8 tablets.
Ingredients
LYNPARZA tablets contain 100 mg or 150 mg of olaparib as the active ingredient and the following inactive ingredients:
- copovidone
- colloidal anhydrous silica
- mannitol
- sodium stearylfumarate
- hypromellose
- macrogol 400
- titanium dioxide
- iron oxide yellow
- iron oxide black (150 mg tablet only)
This medicine does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Manufacturer/Distributor/ Supplier
LYNPARZA is sponsored and supplied in Australia by:
AstraZeneca Pty Ltd
ABN 54 009 682 311
66 Talavera Road
MACQUARIE PARK NSW 2113
Telephone: 1800 805 342
© Copyright 2021
This leaflet was prepared on 26 July 2023
LYNPARZA 100 mg Tablets - AUST R 288613
LYNPARZA 150 mg Tablets - AUST R 288614
Doc ID-003845279 v11
Published by MIMS September 2023



 Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were increased blood creatinine (9%), lymphopenia (6%), VTE (4%), hypersensitivity (2%), MDS/AML (2%), pneumonitis (2%), dermatitis (1%), and increased mean cell volume (0.4%).
Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were increased blood creatinine (9%), lymphopenia (6%), VTE (4%), hypersensitivity (2%), MDS/AML (2%), pneumonitis (2%), dermatitis (1%), and increased mean cell volume (0.4%).
 Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza/bevacizumab were dysgeusia (8%), dyspnoea (8%), stomatitis (5%), dyspepsia (4.3%), erythema (3%), dizziness (2.6%), hypersensitivity (1.7%), and MDS/AML (0.7%).
Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza/bevacizumab were dysgeusia (8%), dyspnoea (8%), stomatitis (5%), dyspepsia (4.3%), erythema (3%), dizziness (2.6%), hypersensitivity (1.7%), and MDS/AML (0.7%).
 Clinically relevant adverse reactions that occurred in < 20% of patients receiving Lynparza were neutropenia (19%), cough (18%), leukopenia (16%), hypomagnesemia (14%), thrombocytopenia (14%), dizziness (13%), dyspepsia (11%), increased creatinine (11%), MDS/AML (8%), oedema (8%), rash (6%), VTE (5%), and lymphopenia (1%).
Clinically relevant adverse reactions that occurred in < 20% of patients receiving Lynparza were neutropenia (19%), cough (18%), leukopenia (16%), hypomagnesemia (14%), thrombocytopenia (14%), dizziness (13%), dyspepsia (11%), increased creatinine (11%), MDS/AML (8%), oedema (8%), rash (6%), VTE (5%), and lymphopenia (1%).
 Clinically relevant adverse reactions that occurred in < 20% of patients receiving Lynparza were dysgeusia (16%), dizziness (15%), dyspnoea (13%), pyrexia (10%), stomatitis (9%), oedema (9%), increase in creatinine (7%), neutropenia (5%), thrombocytopenia (4%), leukopenia (2%), MDS/AML (1%), VTE (1%), and lymphopenia (1%).
Clinically relevant adverse reactions that occurred in < 20% of patients receiving Lynparza were dysgeusia (16%), dizziness (15%), dyspnoea (13%), pyrexia (10%), stomatitis (9%), oedema (9%), increase in creatinine (7%), neutropenia (5%), thrombocytopenia (4%), leukopenia (2%), MDS/AML (1%), VTE (1%), and lymphopenia (1%).
 Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were cough (9.2%), lymphopenia (7%), dyspepsia (6%), upper abdominal pain (4.9%), rash (4.9%), dyspnoea (4.2%), thrombocytopenia (4.2%), increase in creatinine (2%), hypersensitivity (0.9%), VTE (0.5%), dermatitis (0.5%), increase in mean corpuscular volume (0.2%), and MDS/AML (0.2%).
Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were cough (9.2%), lymphopenia (7%), dyspepsia (6%), upper abdominal pain (4.9%), rash (4.9%), dyspnoea (4.2%), thrombocytopenia (4.2%), increase in creatinine (2%), hypersensitivity (0.9%), VTE (0.5%), dermatitis (0.5%), increase in mean corpuscular volume (0.2%), and MDS/AML (0.2%).
 Clinically relevant adverse reactions that occurred in < 20% of patients receiving Lynparza were cough (18%), decreased appetite (16%), thrombocytopenia (11%), dysgeusia (9%), lymphopenia (8%), dyspepsia (8%), dizziness (7%), stomatitis (7%), upper abdominal pain (7%), rash (5%), increase in serum creatinine (3%), dermatitis (1%), and VTE (1%).
Clinically relevant adverse reactions that occurred in < 20% of patients receiving Lynparza were cough (18%), decreased appetite (16%), thrombocytopenia (11%), dysgeusia (9%), lymphopenia (8%), dyspepsia (8%), dizziness (7%), stomatitis (7%), upper abdominal pain (7%), rash (5%), increase in serum creatinine (3%), dermatitis (1%), and VTE (1%).
 Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were cough (9%), abdominal pain upper (7%), blood creatinine increased (7%), dizziness (7%), headache (7%), dyspepsia (5%), leukopenia (5%), VTE (3%), hypersensitivity (2%), and lymphopenia (2%).
Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were cough (9%), abdominal pain upper (7%), blood creatinine increased (7%), dizziness (7%), headache (7%), dyspepsia (5%), leukopenia (5%), VTE (3%), hypersensitivity (2%), and lymphopenia (2%).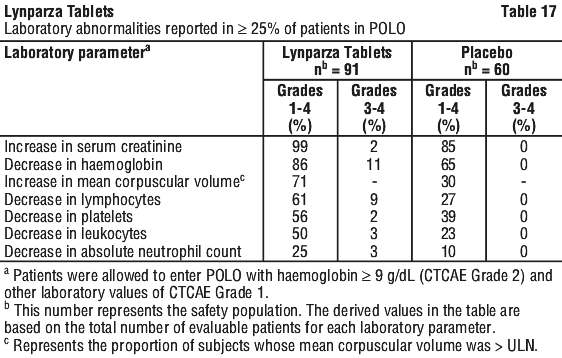
 Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were neutropenia (9%), VTE (7%), dizziness (7%), dysgeusia (7%), dyspepsia (7%), headache (6%), pneumonia (5%), stomatitis (5%), rash (4%), blood creatinine increase (4%), pneumonitis (2%), upper abdominal pain (2%), and hypersensitivity (1%).
Clinically relevant adverse reactions that occurred in < 10% of patients receiving Lynparza were neutropenia (9%), VTE (7%), dizziness (7%), dysgeusia (7%), dyspepsia (7%), headache (6%), pneumonia (5%), stomatitis (5%), rash (4%), blood creatinine increase (4%), pneumonitis (2%), upper abdominal pain (2%), and hypersensitivity (1%).
 Clinically relevant adverse reactions that occurred in < 10% for patients receiving Lynparza plus abiraterone were headache (9%), VTE (8%), rash (7%), dysgeusia (6%), acute kidney injury (3%), and stomatitis (2.5%).
Clinically relevant adverse reactions that occurred in < 10% for patients receiving Lynparza plus abiraterone were headache (9%), VTE (8%), rash (7%), dysgeusia (6%), acute kidney injury (3%), and stomatitis (2.5%).









 Alternatively, for patients who had received prior adjuvant chemotherapy: triple negative breast cancer (TNBC) must have had node-positive disease or node-negative disease with a ≥ 2 cm primary tumour. HR-positive breast cancer must have had ≥ 4 pathologically confirmed positive lymph nodes.
Alternatively, for patients who had received prior adjuvant chemotherapy: triple negative breast cancer (TNBC) must have had node-positive disease or node-negative disease with a ≥ 2 cm primary tumour. HR-positive breast cancer must have had ≥ 4 pathologically confirmed positive lymph nodes.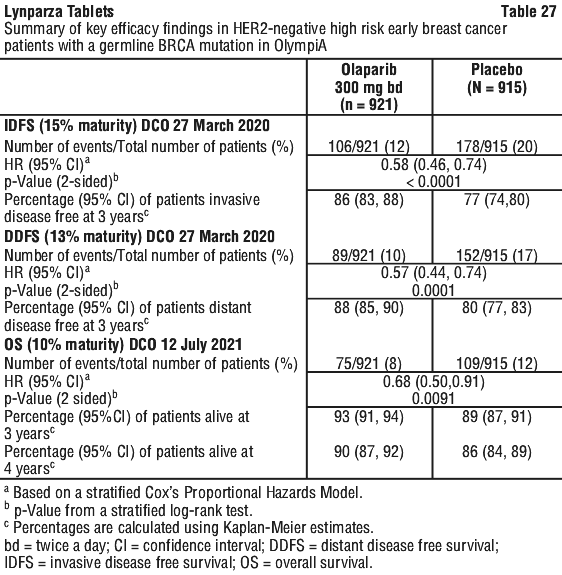
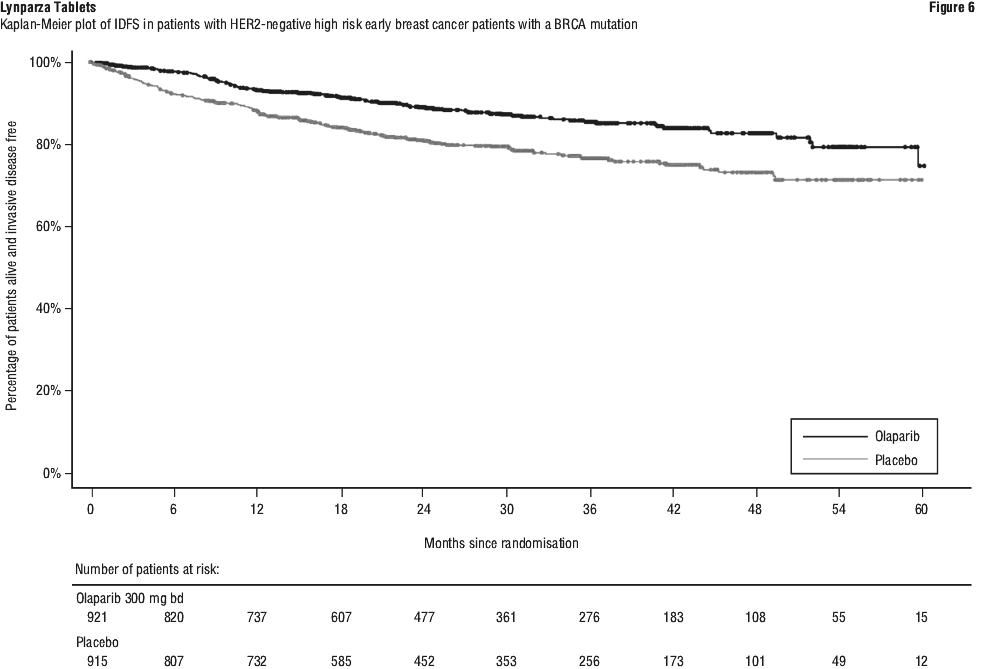



 A significant difference in global health status/QoL (assessed using the EORTC QLQ-C30 questionnaire which uses a 0-100 point scale) in favour of olaparib was observed (adjusted mean difference in change from baseline score was 7.5 points [95% CI: 2.48-12.44; p = 0.0035]). Time to deterioration (≥ 10 points decrease from baseline) in global health status/QoL score was statistically significantly longer on the olaparib arm (HR 0.44; 95% CI: 0.25-0.77; p = 0.0043; median not reached for olaparib vs. 15.3 months for comparator arm).
A significant difference in global health status/QoL (assessed using the EORTC QLQ-C30 questionnaire which uses a 0-100 point scale) in favour of olaparib was observed (adjusted mean difference in change from baseline score was 7.5 points [95% CI: 2.48-12.44; p = 0.0035]). Time to deterioration (≥ 10 points decrease from baseline) in global health status/QoL score was statistically significantly longer on the olaparib arm (HR 0.44; 95% CI: 0.25-0.77; p = 0.0043; median not reached for olaparib vs. 15.3 months for comparator arm).








 Molecular formula: C24H23FN4O3.
Molecular formula: C24H23FN4O3.