
What is in this leaflet
This leaflet answers some common questions about MAVIRET.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor.
Keep this leaflet with the medicine. You may need to read it again.
What MAVIRET is used for
MAVIRET is a tablet that contains two active substances called glecaprevir and pibrentasvir.
This medicine is used to treat chronic (long-lasting) hepatitis C virus (HCV) infection in patients aged 12 years and older.
This medicine belongs to a group of medicines called direct-acting antiviral agents.
This medicine works by stopping the hepatitis C virus from multiplying and infecting new cells. This allows the infection to be permanently eliminated from the body.
Ask your doctor if you have any questions about why it has been prescribed for you. Your doctor may have prescribed it for another purpose.
This medicine is available only with a doctor’s prescription.
This medicine is not expected to affect your ability to drive a car or operate machinery.
Before you take MAVIRET
When you must not take it
Do not take MAVIRET if you have an allergy to:
- any medicine containing glecaprevir or pibrentasvir,
- any of the ingredients listed at the end of this leaflet.
Some symptoms of an allergic reaction include:
- hives, skin rash or itching,
- shortness of breath, wheezing, difficulty breathing or a tight feeling in your chest,
- swelling of the face, lips or tongue, which may cause difficulty in swallowing or breathing.
Do not take MAVIRET if you:
- have severe liver disease.
Do not take MAVIRET if you are taking the following medicines:
- atazanavir (for HIV infection),
- atorvastatin, simvastatin (for lowering cholesterol),
- dabigatran etexilate (for thinning the blood),
- ethinyloestradiol-containing medications (for contraception or hormone replacement therapy),
- rifampicin (for tuberculosis).
Do not give MAVIRET to a child under the age of 12 years. Safety and effectiveness in children younger than 12 years have not been established.
Do not take it after the expiry date printed on the pack or if the packaging is damaged or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
Before you start to take it
Tell your doctor if you have any allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- liver problems other than hepatitis C infection,
- HIV infection,
- hepatitis B infection,
- a liver or kidney transplant
- diabetes.
Tell your doctor or pharmacist if you are pregnant or trying to become pregnant. It is not known if taking MAVIRET while you are pregnant will harm your unborn baby. Your doctor can discuss the risks and benefits involved.
Tell your doctor or pharmacist if you are breastfeeding or are planning to breastfeed. It is not known if MAVIRET can be passed to your baby in your breast milk. Your doctor can discuss the risks and benefits involved.
If you have not told your doctor or pharmacist about any of the above, tell them before you take MAVIRET.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food store.
Keep a list of your medicines to show your doctor or pharmacist.
Do not start taking a new medicine without telling your doctor or pharmacist that you are taking MAVIRET. Some medicines and MAVIRET may interfere with each other. Your doctor or pharmacist can tell you if it is safe to take MAVIRET with other medicines.
Tell your doctor or pharmacist if you are taking any of the following medicines:
- lovastatin, pravastatin or rosuvastatin, medicines used to lower blood cholesterol,
- carbamazepine, used to treat epilepsy,
- ciclosporin, used to suppress the immune system,
- darunavir, ritonavir, efavirenz or lopinavir and ritonavir, used to treat HIV infection,
- digoxin, used to treat heart problems,
- St John's wort (Hypericum perforatum) used to treat mild depression,
- Vitamin K antagonist, for example warfarin, used to thin the blood.
These medicines may be affected by MAVIRET, or may affect how well it works. You may need to use different amounts of your medicine, or take different medicines.
Your doctor or pharmacist has more information on medicines to be careful with or to avoid while taking MAVIRET.
How to take MAVIRET
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
It is important you follow the instructions your doctor provides about how to take MAVIRET and for how long.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
Not taking the medicine as directed or stopping the medicine too early may cause you to not respond to the medicine and may affect your response to future treatments.
How much to take and when to take it
MAVIRET tablets are taken once a day with food.
Patients aged 12 years and older:
Take 3 tablets.
When to take it
Take MAVIRET at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How to take it
Swallow the tablets whole with a full glass of water.
Do not crush, chew or break the tablets.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
Do not stop taking MAVIRET without first talking with your doctor.
It is important to keep taking your medicine even if you feel well.
If you forget to take it
It is important not to miss a dose of this medicine. If you miss a dose, and it is:
- less than 18 hours from the time you usually take MAVIRET, take the missed dose with food as soon as possible. Then take your next dose at your usual time.
- more than 18 hours from the time you usually take MAVIRET, do not take the missed dose. Take your next dose as usual with food.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for advice.
If you take too much (overdose)
Immediately telephone your doctor, or the Poisons Information Centre (telephone Australia 13 11 26), or go to Accident and Emergency at your nearest hospital, if you think you or anyone else may have taken too much MAVIRET. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using MAVIRET
Things you must do
If you are about to start on any new medicine, remind your doctor and pharmacist that you are taking MAVIRET.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon that you are taking this medicine.
If you become pregnant while you are taking this medicine, tell your doctor or pharmacist immediately.
Keep all of your doctor’s appointments so that your progress can be checked.
Things you must not do
Do not use this medicine to treat any other complaints unless your doctor or pharmacist tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not stop taking MAVIRET, or change the dosage, without checking with your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking MAVIRET.
All medicines have some unwanted side effects. Sometimes they are serious, but most of the time they arenot. You may need medical attention if you get some of the side-effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- feeling very tired,
- headache,
- nausea.
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor or pharmacist if you notice the following and it worries you:
- itching (called pruritus)
The above list includes a side effect of unknown frequency.
Tell your doctor immediately, or go to Accident and Emergency at your nearest hospital if you notice the following:
- swelling of the face, lips, tongue or throat (called angioedema)
The above list includes a side effect of unknown frequency.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may occur in some people.
After using MAVIRET
Storage
Keep your tablets in the carton or bottle until it is time to take them.
Keep the medicine in a cool, dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom, near a sink, or on a windowsill. Do not leave it in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and- a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine, or the medicine has passed its expiry date, ask your pharmacist what todo with any medicine that is left over.
Product description
What it looks like
MAVIRET tablets are pink, oblong, film-coated tablets that are curved on both sides, and debossed on one side with 'NXT'.
MAVIRET is available in a carton or bottle containing 84 tablets.
Ingredients
MAVIRET contains 100 mg of glecaprevir and 40 mg of pibrentasvir as active ingredients.
It also contains:
- copovidone,
- tocofersolan,
- colloidal anhydrous silica,
- propylene glycol monocaprylate,
- croscarmellose sodium,
- sodium stearyl fumarate,
- hypromellose 2910,
- lactose monohydrate,
- titanium dioxide,
- macrogol 3350,
- iron oxide red.
MAVIRET contains lactose.
MAVIRET does not contain gluten.
Supplier
MAVIRET is supplied in Australia by:
AbbVie Pty Ltd
241 O'Riordan Street
Mascot NSW 2020 Australia
This leaflet was prepared on 24 December 2019.
Australian Registration Number(s)
AUST R 284948
AUST R 325401
Version No. 6
Published by MIMS March 2020


 The adult and adolescent dose of Maviret tablets should be used in children weighing 45 kg or greater (see Recommended dosage of Maviret tablets in adults and adolescents 12 years and older or weighing at least 45 kg).
The adult and adolescent dose of Maviret tablets should be used in children weighing 45 kg or greater (see Recommended dosage of Maviret tablets in adults and adolescents 12 years and older or weighing at least 45 kg).


 The EC50 values of glecaprevir and pibrentasvir against chimeric replicons encoding NS3 or NS5A from clinical isolates are presented in Table 8.
The EC50 values of glecaprevir and pibrentasvir against chimeric replicons encoding NS3 or NS5A from clinical isolates are presented in Table 8.
 Serum HCV RNA values were measured during the clinical studies using the Roche COBAS AmpliPrep/COBAS TaqMan HCV test (version 2.0) with a lower limit of quantification (LLOQ) of 15 IU/mL (except for SURVEYOR-1 and SURVEYOR-2 which used the Roche COBAS TaqMan real-time reverse transcriptase-PCR (RT-PCR) assay v. 2.0 with an LLOQ of 25 IU/mL). Sustained virologic response (SVR12), defined as HCV RNA less than LLOQ at 12 weeks after the cessation of treatment, was the primary endpoint in all the studies to determine the HCV cure rate.
Serum HCV RNA values were measured during the clinical studies using the Roche COBAS AmpliPrep/COBAS TaqMan HCV test (version 2.0) with a lower limit of quantification (LLOQ) of 15 IU/mL (except for SURVEYOR-1 and SURVEYOR-2 which used the Roche COBAS TaqMan real-time reverse transcriptase-PCR (RT-PCR) assay v. 2.0 with an LLOQ of 25 IU/mL). Sustained virologic response (SVR12), defined as HCV RNA less than LLOQ at 12 weeks after the cessation of treatment, was the primary endpoint in all the studies to determine the HCV cure rate. Of the GT1, 2, 4, 5 or 6-infected patients with end stage renal disease enrolled in EXPEDITION-4, 97.8% (91/93) achieved SVR12 with no virologic failures.
Of the GT1, 2, 4, 5 or 6-infected patients with end stage renal disease enrolled in EXPEDITION-4, 97.8% (91/93) achieved SVR12 with no virologic failures.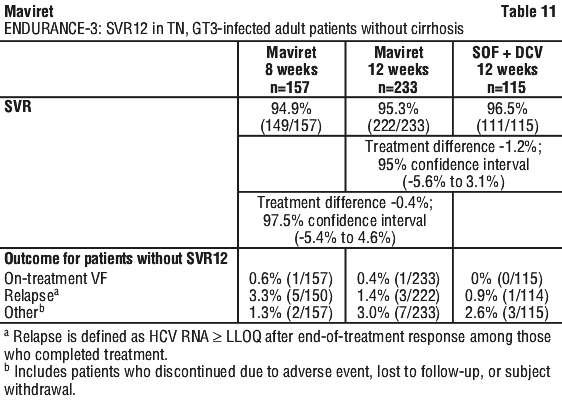
 Of the GT3-infected patients with end stage renal disease enrolled in EXPEDITION-4, 100% (11/11) achieved SVR12.
Of the GT3-infected patients with end stage renal disease enrolled in EXPEDITION-4, 100% (11/11) achieved SVR12. Lower SVR12 rates were observed in GT1a-infected patients who were retreated with Maviret within 12 months of failing a regimen containing both NS3/4A protease inhibitors and NS5A inhibitors.
Lower SVR12 rates were observed in GT1a-infected patients who were retreated with Maviret within 12 months of failing a regimen containing both NS3/4A protease inhibitors and NS5A inhibitors.
 Relative to healthy subjects (N = 230), glecaprevir Cmax was 51% lower and AUC24,ss was similar (10% difference) in HCV-infected patients without cirrhosis; pibrentasvir Cmax and AUC24,ss were 63% and 34% lower, respectively.
Relative to healthy subjects (N = 230), glecaprevir Cmax was 51% lower and AUC24,ss was similar (10% difference) in HCV-infected patients without cirrhosis; pibrentasvir Cmax and AUC24,ss were 63% and 34% lower, respectively.



