What is in this leaflet
Please read this leaflet carefully before you start taking MEKTOVI
This leaflet answers some common questions about MEKTOVI. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking MEKTOVI against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What MEKTOVI is used for
MEKTOVI is an anti-cancer medicine containing the active ingredient binimetinib, which belongs to a group of medicines called ‘MEK inhibitors’.
MEKTOVI is used in combination with another medicine which contains the active ingredient, encorafenib (called BRAFTOVI®) to treat adult patients with a type of skin cancer called melanoma, which has spread to other parts of the body, or cannot be removed by surgery. The type of melanoma which MEKTOVI and BRAFTOVI are used to treat, has a particular change (mutation) in a gene called BRAF. This mutation in the BRAF gene may have produced proteins which caused the melanoma to develop.
Before you start treatment, your doctor will have tested you to confirm that you have this BRAF mutation.
MEKTOVI targets a protein called MEK which promotes cancer cell growth. When MEKTOVI is used in combination with BRAFTOVI (which targets the changed BRAF protein), it further slows down or stops the growth of your cancer.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
BRAFTOVI is not recommended for children and adolescents aged under 18 years. The safety and efficacy of this medicine has not been established in this age group.
Before you take MEKTOVI
As MEKTOVI is to be used in combination with BRAFTOVI, you must also read the Consumer Medicine Information for BRAFTOVI.
When you must not take it
Do not take MEKTOVI if you have an allergy to:
- any medicine containing binimetinib
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
If you experience an allergic reaction, stop taking the medicine and inform your doctor or pharmacist immediately.
Do not take MEKTOVI after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. The expiry date refers to the last day of that month. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor, nurse or pharmacist if you have allergies to any other medicines, foods, preservatives or dyes.
MEKTOVI contains lactose. Tell your doctor, nurse or pharmacist if you have an intolerance to some sugars.
Tell your doctor if you have or have had any of the following medical conditions:
- heart problems
- high blood pressure
- muscle problems
- blood clots
- liver problems
- lung or breathing problems
- bleeding problems or if you are taking medicines that may increase your risk of bleeding
- eye problems including glaucoma or increased pressure in your eyes
Tell your doctor if you have had a history of blockage in the vein draining the eye (retinal vein occlusion) as MEKTOVI is not recommended in patients with a history of retinal vein occlusion.
Tell your doctor if you have had a different type of cancer than melanoma as MEKTOVI when taken with BRAFTOVI may cause progression of certain other types of cancers.
Pregnancy
Taking MEKTOVI during pregnancy is not recommended. Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding before taking MEKTOVI.
MEKTOVI may cause permanent harm or birth defects to an unborn baby.
Breast-feeding
MEKTOVI is not recommended while breast-feeding. If you are breastfeeding or planning to breastfeed, you must tell your doctor before taking this medicine.
It is not known if MEKTOVI passes into breastmilk.
If you have not told your doctor about any of the above, tell him/her before you start taking MEKTOVI.
Taking other medicines
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
These medicines may be affected by MEKTOVI or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Keep a list of the medicines you take so you can show it to your doctor, nurse or pharmacist when you get a new medicine
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take MEKTOVI
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions in this leaflet, ask your doctor or pharmacist for help.
How much to take
Always take MEKTOVI exactly as your doctor has prescribed.
The recommended dose of MEKTOVI, when taken in combination with BRAFTOVI, is three 15 mg tablets twice daily taken about 12 hours apart (corresponding to a daily dose of 90 mg).
If you experience serious side effects (such as skin, eye, heart or lung problems), your doctor may lower the dose of MEKTOVI, or stop treatment temporarily or permanently.
How to take it
Swallow the tablets whole with a full glass of water.
MEKTOVI can be taken with or without food.
If vomiting occurs at any time after taking the tablets do not take an additional dose. Take the next dose as scheduled.
How long to take it
Continue taking MEKTOVI for as long as your doctor tells you to. Do not stop unless your doctor advises you to.
If you forget to take it
If you miss a dose of MEKTOVI:
- If the missed dose is less than 6 hours late, take it as soon as you remember;
- If the missed dose is more than 6 hours late, skip that dose and take your next dose at the usual time.
Then go back to taking your tablets as you would normally.
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your doctor, nurse or pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (In Australia telephone 13 11 26. In New Zealand telephone 0800 764 766) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much MEKTOVI. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking MEKTOVI
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking MEKTOVI.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
If you become pregnant while taking MEKTOVI, tell your doctor immediately.
If you are a woman who could become pregnant, you must use effective birth control (contraception) while you are taking MEKTOVI and you must continue to use effective contraception for at least 1 month after taking your last dose.
Tell your doctor if you are breastfeeding while being treated with MEKTOVI.
Tell your doctor, nurse or pharmacist immediately if you experience the following while you are taking MEKTOVI:
Skin changes
MEKTOVI when taken with BRAFTOVI may cause other types of skin cancer such as cutaneous squamous cell carcinoma.
Your doctor will periodically check for new cancers on your skin and inside your body before, during and after your treatment. Tell your doctor immediately if you detect any skin changes including new warts, skin soreness, reddish bumps which bleed or don’t heal or any changes in the size or colour of a mole.
Heart problems
MEKTOVI can lower the amount of blood pumped by your heart or make existing heart problems worse. Your doctor will run tests to check that your heart is working properly before and during your treatment with this medicine.
Blood clots
MEKTOVI can cause blood clots in your arms or legs which can travel to your lungs and lead to death. If necessary, your doctor may decide to interrupt or completely stop your MEKTOVI treatment.
Bleeding problems
MEKTOVI may cause serious bleeding problems. Tell your doctor immediately if you have any signs of bleeding.
Eye problems
MEKTOVI can cause serious eye problems. Your doctor will examine your eyes for any new or worsening problems with your sight while you are taking this medicine.
Liver problems
MEKTOVI can increase the amounts of liver enzymes in your blood. Your doctor will run blood tests to monitor your liver function before and during treatment.
Muscle problems
MEKTOVI can cause breakdown of muscle (rhabdomyolysis). Your doctor will run blood tests to monitor muscle condition before and during treatment. As a precaution, drink plenty of fluids during treatment, unless otherwise advised by your doctor.
High blood pressure
MEKTOVI can raise blood pressure. Your doctor or nurse will check your blood pressure before and during treatment with MEKTOVI.
Lung or breathing problems
MEKTOVI may cause lung or breathing problems including inflammation of the lungs (pneumonitis or interstitial lung disease). Signs and symptoms can include: cough, shortness of breath or fatigue. If necessary, your doctor may interrupt or completely stop your MEKTOVI treatment.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not take Mektovi to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if their symptoms seem similar to yours or they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
MEKTOVI can affect your ability to drive or use machines.
Be careful driving or operating machinery until you know how MEKTOVI affects you. If you experience any problems with your vision, or any other side-effects that may affect your ability, avoid driving or using machines. Talk to your doctor if you are not sure if you should drive.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking MEKTOVI.
Like other medicines, MEKTOVI can cause side effects but not everybody gets them. Some may be serious and need medical attention.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Possible serious side effects
MEKTOVI used in combination with BRAFTOVI may cause serious side effects. Tell your doctor immediately if you experience any of the following serious side effects, either for the first time or if they get worse.
Heart problems
MEKTOVI can affect how well your heart pumps (left ventricular dysfunction). Signs and symptoms can include:
- feeling dizzy, tired or lightheaded
- shortness of breath
- feeling like your heart is pounding, racing or beating irregularly
- swelling in the legs
High blood pressure
MEKTOVI can increase blood pressure. Tell your doctor immediately if you experience severe headache, feel dizzy or lightheaded, or if your blood pressure is much higher than usual (if you are self-monitoring your blood pressure at home).
Blood clots
MEKTOVI may cause blood clots (venous thromboembolism including pulmonary embolism). Signs and symptoms can include:
- chest pain
- sudden shortness of breath or trouble breathing
- pain in your legs with or without swelling
- swelling in your arms and legs
- a cool, pale arm or leg
Eye problems
MEKTOVI may induce fluid leakage under the retina in the eye that results in detachment of different layers in the eye (retinal pigment epithelial detachment), which could lead to the following symptoms:
- blurred vision, loss of vision or other vision changes (e.g. coloured dots in your vision)
- halo (seeing blurred outline around objects)
- eye pain, swelling or redness
Muscle problems
MEKTOVI may lead to breakdown of muscles (rhabdomyolysis) which can lead to kidney damage and can be fatal. Signs and symptoms can include:
- muscle pain, cramps, stiffness or spasm
- dark urine
Bleeding problems
Taking MEKTOVI can cause serious bleeding problems. Tell your doctor immediately if you have any unusual bleeding or signs of bleeding including:
- headaches, dizziness or weakness
- coughing up of blood or blood clots
- vomit containing blood or that looks like “coffee grounds”
- red or black tools that look like tar
- passing blood in the urine
- stomach (abdominal) pain
- unusual vaginal bleeding
Other skin cancers
MEKTOVI when taken with BRAFTOVI may cause other types of skin cancer such as cutaneous squamous cell carcinoma. Usually these skin cancers can be removed with surgery and treatment with MEKTOVI and BRAFTOVI can be continued without interruption.
Other side effects
Besides the serious side effects mentioned above, taking MEKTOVI in combination with BRAFTOVI may cause other side effects.
When MEKTOVI was taken with BRAFTOVI the following side effects were reported.
Very common side effects (may affect more than 1 in 10 people)
- reduced red blood cell count (anaemia)
- problems with nerves that can cause pain, loss of sensation or tingling in hands and feet
- headache
- dizziness
- bleeding at various sites in the body
- problems with your vision (visual impairment)
- stomach pain
- diarrhoea
- being sick (vomiting)
- feeling sick (nausea)
- constipation
- itching
- dry skin
- abnormal hair loss or thinning (alopecia)
- skin rash of various types
- thickening of the outer layers of the skin
- joint pain (arthralgia)
- muscle pain (myalgia), weakness or spasm
- back pain
- pain in the hands and feet
- fever
- swelling of the hands or feet (peripheral oedema), localised swelling
- fatigue
- abnormal blood test results for liver
- abnormal blood test results related to blood creatine kinase, indicating damage to heart and muscle
Common side effects (may affect up to 1 in 10 people)
- some types of skin tumours such as skin papilloma and basal cell carcinoma
- allergic reaction that may include swelling of the face and difficulty breathing
- changes in the way things taste
- inflammation of the eye (uveitis)
- inflammation of the colon (colitis)
- redness, chapping or cracking of the skin
- inflammation of the fatty layer under the skin, symptoms include tender skin nodules
- skin rash with a flat discoloured area or raised bumps like acne (dermatitis acneiform)
- redness, skin peeling or blisters on the hands and feet (palmar-plantar erythrodysesthesia or hand and foot syndrome)
- kidney failure
- abnormal kidney test results (creatinine elevations)
- abnormal blood test results for liver function (blood alkaline phosphatase)
- abnormal blood test results for pancreas function (amylase, lipase)
- increased skin sensitivity to sunlight
- Uncommon side effects (may affect up to 1 in 100 people)
- weakness and paralysis of the face muscles (facial paresis)
- inflammation of the pancreas (pancreatitis) causing severe abdominal pain
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After using MEKTOVI
Storage
Keep your MEKTOVI tablets in a place where the temperature stays below 30°C.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Do not store MEKTOVI or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Do not throw any medicines away via wastewater or household waste.
Product description
What it looks like
MEKTOVI 15 mg tablets are supplied in blister packs of 84 tablets (7 strips of 12 tablets).
The 15 mg tablets are yellow/dark yellow, unscored biconvex, oval and film-coated, with “A” debossed on one face and “15” on the opposite face..
Ingredients
MEKTOVI contains 15 mg of binimetinib as the active ingredient.
Tablet core:
- lactose monohydrate
- microcrystalline cellulose
- colloidal anhydrous silica
- croscarmellose sodium
- magnesium stearate
Tablet coating:
- polyvinyl alcohol
- macrogol 3350
- titanium dioxide
- purified talc
- iron oxide yellow
- iron oxide black
Supplier
MEKTOVI is supplied in Australia by:
Pierre Fabre Australia Pty Limited
901/1 Elizabeth Plaza
North Sydney NSW 2060
Australia
This leaflet was prepared in May 2019.
AUSTR number: AUST R 295440
Internal document code
(Aus Mektovi CMI v4 based on Aus Mektovi PI v7)
Published by MIMS August 2019



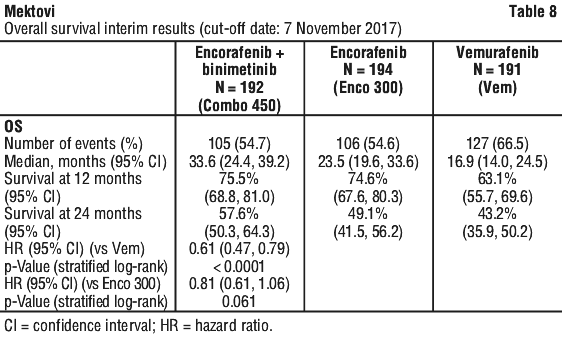
 In Part 1 of study CMEK162B2301, serious adverse events (SAEs) regardless of relationship to study therapy were reported in 35.9% of patients treated with encorafenib 450 mg in combination with binimetinib (Combo 450), 34.9% of patients treated with encorafenib single agent 300 mg (Enco 300) and in 38.2% of patients treated with vemurafenib.
In Part 1 of study CMEK162B2301, serious adverse events (SAEs) regardless of relationship to study therapy were reported in 35.9% of patients treated with encorafenib 450 mg in combination with binimetinib (Combo 450), 34.9% of patients treated with encorafenib single agent 300 mg (Enco 300) and in 38.2% of patients treated with vemurafenib.

 The efficacy results based on investigator assessment were consistent with the independent central assessment. The results by investigator assessment are summarised in Table 7.
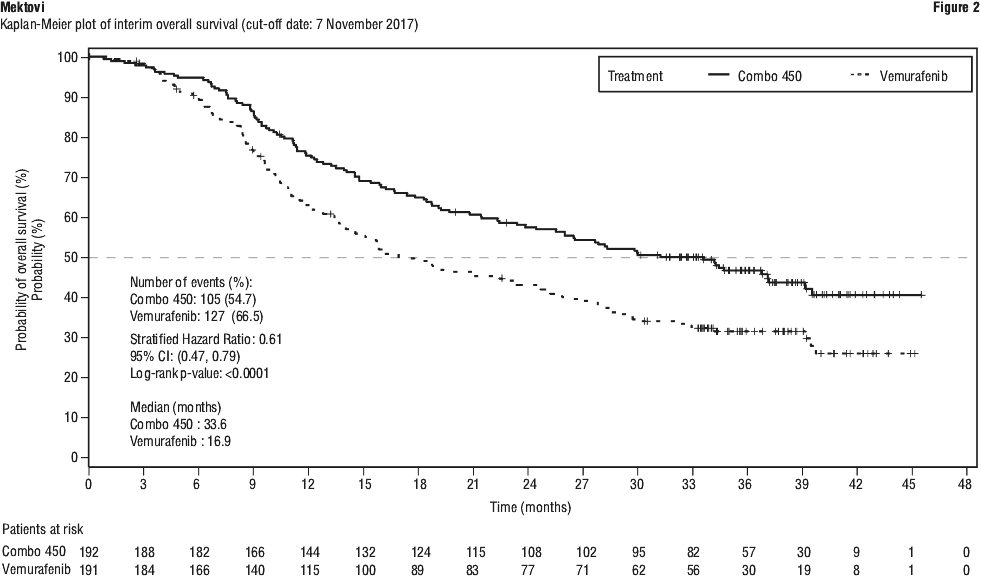
The efficacy results based on investigator assessment were consistent with the independent central assessment. The results by investigator assessment are summarised in Table 7.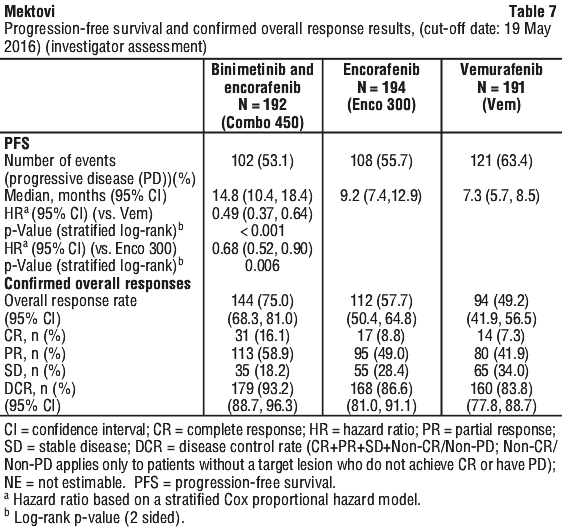 At a cut-off date of 07 November 2017, an update of the PFS analyses was performed. The PFS analysis per independent central assessment showed an improvement of PFS in patients treated with Combo 450 compared with patients treated with vemurafenib (14.9 vs 7.3 months, respectively), HR 0.51 (95% CI: 0.39, 0.67) (p < 0.001 one sided) and also compared with patients treated with encorafenib (14.9 vs 9.6 months, respectively), HR 0.77 (95% CI: 0.59, 1.0) (p = 0.0249 one sided). The analysis per independent central assessment showed that encorafenib improved PFS vs. vemurafenib (9.6 vs 7.3 months, respectively), HR 0.68 (95% CI: 0.52, 0.88) (p = 0.0019 one sided).
At a cut-off date of 07 November 2017, an update of the PFS analyses was performed. The PFS analysis per independent central assessment showed an improvement of PFS in patients treated with Combo 450 compared with patients treated with vemurafenib (14.9 vs 7.3 months, respectively), HR 0.51 (95% CI: 0.39, 0.67) (p < 0.001 one sided) and also compared with patients treated with encorafenib (14.9 vs 9.6 months, respectively), HR 0.77 (95% CI: 0.59, 1.0) (p = 0.0249 one sided). The analysis per independent central assessment showed that encorafenib improved PFS vs. vemurafenib (9.6 vs 7.3 months, respectively), HR 0.68 (95% CI: 0.52, 0.88) (p = 0.0019 one sided).