1 Name of Medicine
Tenecteplase (rch).
2 Qualitative and Quantitative Composition
Metalyse 25 mg.
1 vial contains 25 mg (5,000 units) tenecteplase.
The reconstituted solution contains 5 mg (1,000 units) tenecteplase per mL.
The potency of Metalyse expressed in Units (U) is based on a reference standard that is specific for tenecteplase. The U for tenecteplase is not comparable with units used for other thrombolytic agents.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Metalyse is a sterile, white to off-white, lyophilised powder for single intravenous bolus administration after reconstitution with sterile water for injections.
4.1 Therapeutic Indications
Metalyse is indicated in adults for the thrombolytic treatment of acute ischaemic stroke (AIS) within 4.5 hours from last known well and after exclusion of intracranial haemorrhage.
4.2 Dose and Method of Administration
The appropriate presentation of tenecteplase product should be chosen carefully and in line with the indication. Metalyse 25 mg is intended for use in acute ischaemic stroke only.
Metalyse should be administered as early as possible and no later than 4.5 hours after last known well and after exclusion of intracranial haemorrhage by appropriate imaging techniques (see Section 4.4 Special Warnings and Precautions for Use). The treatment effect is time-dependent; therefore, earlier treatment increases the probability of a favourable outcome.
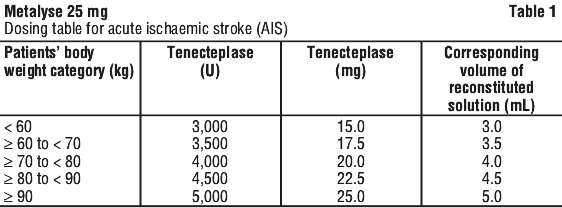
Metalyse should be administered on the basis of body weight, with a maximum single dose of 25 mg (5,000 U) tenecteplase.
Benefit-risk of tenecteplase treatment should be carefully evaluated in patients weighing 50 kg or less due to limited availability of data.
The doses for different weight categories given in Table 1 are based on a tenecteplase dose of 0.25 mg/kg (maximum dose 25 mg).
The volume required to administer the correct total dose can be calculated from Table 1.
Adjunctive therapy.
The safety and efficacy of this regimen with concomitant treatment with heparin (unfractionated or low molecular weight) or platelet aggregation inhibitors such as aspirin during the first 24 hours after treatment with Metalyse have not been investigated sufficiently. Therefore, administration of heparin or platelet aggregation inhibitors such as aspirin should be avoided in the first 24 hours after treatment with Metalyse due to an increased haemorrhagic risk.
Any concomitant use of anticoagulants and/or antiplatelets should be based on clinical guidelines and individual risk-benefit assessment.
Method of administration.
The reconstituted solution should be administered intravenously and is for immediate use.
The required dose should be administered as a single intravenous bolus over 5 to 10 seconds.
The reconstituted solution is for single use, in a single patient. Any excess solution should be discarded.
Reconstitution and handling.
Metalyse should be reconstituted by adding 5 mL of sterile water for injections to the vial containing the powder for solution for injection using a needle and a syringe (not provided in the package).
1. Remove the crimp cap from the vial.
2. Fill a syringe with 5 mL of sterile water for injections and penetrate the vial stopper in the middle with the needle.
3. Add all the sterile water for injections into the vial by pushing the syringe plunger down slowly to avoid foaming.
4. Keep the syringe attached to the vial and reconstitute by swirling gently.
5. The reconstituted preparation is a colourless to pale yellow, clear solution. Only clear solution without particles should be used.
6. Directly before the solution is administered, invert the vial with the syringe still attached, so that the syringe is below the vial.
7. Transfer the appropriate volume of reconstituted solution of Metalyse into the syringe, based on the patient's weight (see Table 1).
8. A pre-existing intravenous line, which has been used for administration of 0.9% sodium chloride solution only, may be used for administration of Metalyse. Metalyse should not be mixed with other drugs, neither in the same vial nor the same venous line.
9. Metalyse should be administered as a single dose to the patient, intravenously over 5 to 10 seconds. It should not be administered into a line containing dextrose as Metalyse is incompatible with dextrose solution.
10. The line should be flushed after Metalyse injection for proper delivery.
11. Any unused solution should be discarded.4.3 Contraindications
Metalyse is contraindicated in:
Acute ischaemic stroke without disabling neurological deficit.
Situations associated with a risk of bleeding such as:
history or evidence or suspicion of intracranial haemorrhage including subarachnoid haemorrhage;
significant bleeding disorder at present or within the past 6 months, known haemorrhagic diathesis;
active systemic non-compressible bleeding;
patients with effective anticoagulation (e.g. INR > 1.7) (please see Section 4.4 Special Warnings and Precautions for Use, Bleeding);
any history of central nervous system damage (i.e. neoplasm, aneurysm, intracranial or spinal surgery);
thoracic aortic dissection;
severe uncontrolled arterial hypertension (see Section 4.4 Special Warnings and Precautions for Use, Blood pressure monitoring);
prolonged or traumatic cardiopulmonary resuscitation (> 2 minutes) within the past 2 weeks;
severe hepatic dysfunction, including hepatic failure, cirrhosis, portal hypertension (oesophageal varices) and active hepatitis;
active peptic ulceration;
arterial aneurysm and known arterial/venous malformation;
neoplasm with increased bleeding risk;
acute pericarditis and/or subacute bacterial endocarditis;
acute pancreatitis.
Patients with known hypersensitivity to the active substance tenecteplase, gentamicin (a trace residue from the manufacturing process) or to any of the excipients.
4.4 Special Warnings and Precautions for Use
The appropriate presentation of tenecteplase product should be chosen carefully and in line with the indication. Metalyse 25 mg is intended for use in acute ischaemic stroke only.
Metalyse should be prescribed by physicians experienced in the use of thrombolytic treatment and with the facilities to monitor that use. This does not preclude the pre-hospital use of Metalyse. As with other thrombolytics, it is recommended that when Metalyse is administered standard resuscitation equipment and medication be available in all circumstances.
Treatment must be performed with the involvement of physicians trained and experienced in neurological care. For the indication verification, remote diagnostic measures may be considered as appropriate (see Section 4.2 Dose and Method of Administration).
Traceability.
In order to improve traceability of biological medicinal products, the trade name and the batch number of the administered product should be clearly recorded in the patient file.
Bleeding.
The most common complication encountered during Metalyse therapy is bleeding.
The concomitant use of other active substances affecting coagulation or platelet function (e.g. heparin) may contribute to bleeding (see Section 4.3 Contraindications).
Should serious bleeding occur, in particular cerebral haemorrhage, concomitant administration of such active substances should be terminated immediately. Administration of protamine should be considered if heparin has been administered within 4 hours before the onset of bleeding. In the few patients who fail to respond to these conservative measures, judicious use of transfusion products may be indicated. Transfusion of cryoprecipitate, fresh frozen plasma, and platelets should be considered with clinical and laboratory reassessment after each administration. A target fibrinogen level of 1 g/L is desirable with cryoprecipitate infusion. Antifibrinolytic agents should also be considered.
As fibrin is lysed during Metalyse therapy, bleeding from recent puncture sites may occur. Therefore, thrombolytic therapy requires careful attention to all possible bleeding sites (including those following catheter insertions, arterial and venous puncture, cutdown and needle puncture). The use of rigid catheters, intramuscular injections and non-essential handling of the patient should be avoided during treatment with Metalyse.
The use of Metalyse therapy has to be carefully evaluated in order to balance the potential risks of bleeding with expected benefits under the following conditions:
Low body weight < 60 kg.
Patients receiving oral anticoagulants treatment: the use of Metalyse may be considered when appropriate test(s) show no clinically relevant anticoagulant activity.
Recent intramuscular injection or small recent traumas, such as biopsies, puncture of major vessels, cardiac massage for resuscitation.
Extensive infarctions (e.g. NIHSS > 25).
Recent history of previous stroke or serious head or spinal trauma or major surgery (such as cardiac, thoracic, abdominal, or orthopaedic).
Elevated activated partial thromboplastin time (aPTT) at presentation.
Platelet count of less than 100,000/mm3.
Ischaemic stroke or transient ischaemic attack (TIA) in the preceding 6 months.
Recent (within 10 days) gastrointestinal or genitourinary bleeding.
Recent (within 10 days) obstetrical delivery, organ biopsy, puncture of non-compressible blood vessel (e.g. subclavian or jugular vein puncture).
Haemostatic defects including those secondary to severe hepatic or renal disease; special attention should be paid to coagulation parameters in patients with significant liver dysfunction.
Intracerebral haemorrhages represent the most frequent adverse event. However, this did not result in an increased overall morbidity or mortality.
The risk of intracranial haemorrhage in acute ischaemic stroke patients may be increased with the use of Metalyse.
This applies in particular in the following cases:
All situations involving a high risk of haemorrhage including those listed in Section 4.3 Contraindications.
Late time-to-treatment onset.
Patients pre-treated with aspirin may have a greater risk of intracerebral haemorrhage, particularly if Metalyse treatment is delayed.
Compared to younger patients, patients of advanced age (over 80 years) may have a somewhat poorer outcome independent of treatment and may have an increased risk of intracerebral haemorrhage when thrombolysed. In general, the benefit-risk of thrombolysis in patients of advanced age remains positive. Thrombolysis in AIS patients should be evaluated on individual benefit-risk basis.
Treatment must not be initiated later than 4.5 hours after last known well because of unfavourable benefit/risk ratio mainly based on the following:
positive treatment effects decrease over time;
the mortality rate increases particularly in patients with prior aspirin treatment;
risk of symptomatic haemorrhage increases.
Hypersensitivity.
Immune-mediated hypersensitivity reactions associated with the administration of Metalyse can be caused by the active substance tenecteplase, gentamicin (a trace residue from the manufacturing process) or any of the excipients (also see Section 4.3 Contraindications).
No sustained antibody formation to the tenecteplase molecule has been observed after treatment. However, there is no experience with re-administration of Metalyse.
There is also a risk of hypersensitivity reactions mediated through a non-immunological mechanism.
Angio-oedema represents the most common hypersensitivity reaction reported with Metalyse. This risk may be enhanced in the indication acute ischaemic stroke and/or by concomitant treatment with ACE inhibitors.
Patients treated with Metalyse should be monitored for angio-oedema during and for up to 24 h after administration.
If a severe hypersensitivity reaction (e.g. angio-oedema) occurs, appropriate treatment should be promptly initiated. This may include intubation.
Cholesterol embolisation.
Cholesterol embolism has been reported rarely in patients treated with all types of thrombolytic agents; the true incidence is unknown. This serious condition, which can be lethal, is also associated with invasive vascular procedures (e.g. cardiac catheterisation, angiography, vascular surgery) and/or anticoagulant therapy. Clinical features of cholesterol embolism may include livedo reticularis, "purple toe" syndrome, acute renal failure, gangrenous digits, hypertension, pancreatitis, myocardial infarction, cerebral infarction, spinal cord infarction, retinal artery occlusion, bowel infarction, and rhabdomyolysis.
Thrombo-embolism.
The use of Metalyse can increase the risk of thrombo-embolic events in patients with left heart thrombus, e.g. mitral stenosis or atrial fibrillation.
Blood pressure monitoring.
To initiate thrombolysis, uncontrolled hypertension or systolic blood pressure (BP) > 185 mmHg or diastolic BP > 110 mmHg should be carefully managed.
BP monitoring during the first 24 hours is necessary. Intravenous antihypertensive therapy is recommended if systolic BP > 180 mmHg or diastolic BP > 105 mmHg.
Special patient groups at reduced benefit-risk.
The benefit/risk ratio of thrombolytic therapy is considered less favourable in patients who have had a prior stroke or in whom uncontrolled diabetes exists, although still positive in these patients.
The benefit/risk ratio of Metalyse administration should be thoroughly considered in AIS patients with the following conditions:
rapidly improving symptoms;
seizure at the onset of stroke;
blood glucose < 2.7 mmol/L or > 22.2 mmol/L, which must be corrected before treatment initiation.
In stroke patients, the likelihood of a favourable outcome decreases with longer time from last known well to thrombolytic treatment, increasing age, increasing stroke severity and increased levels of blood glucose on admission while the likelihood of severe disability and death or symptomatic intracranial bleeding increases, independently of treatment.
Cerebral oedema.
Reperfusion of the ischaemic area may induce cerebral oedema in the infarcted zone.
Use in the elderly.
Metalyse should be administered with caution in the elderly (> 80 years) due to a higher bleeding risk (see Section 4.4 Special Warnings and Precautions for Use, Bleeding).
Paediatric Use.
Safety and efficacy data in children below 18 years of age are not available for Metalyse. Therefore, Metalyse is not recommended for use in children below 18 years of age.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No formal interaction studies with Metalyse and medicinal products commonly administered in patients with acute ischaemic stroke have been performed.
Drugs affecting coagulation/platelet function.
Medicinal products that affect coagulation or those that alter platelet function may increase the risk of bleeding, and should therefore be avoided in the first 24 hours after Metalyse treatment for acute ischaemic stroke (see Section 4.3 Contraindications).
ACE inhibitors.
Concomitant treatment with ACE inhibitors may enhance the risk of experiencing a hypersensitivity reaction (see Section 4.4 Special Warnings and Precautions for Use).4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Clinical data as well as nonclinical studies on fertility are not available for tenecteplase.
(Category C)
Thrombolytic agents can produce placental haemorrhage and subsequent prematurity and fetal loss.
Treatment of rabbits with tenecteplase 0.5-5 mg/kg/day during mid gestation caused vaginal haemorrhage and subsequent embryonic death (approximately 0.8 times maximum clinical exposure, based on AUC). A no effect dose was not established. No fetal abnormalities were detected. There were no adverse effects on the pregnant animal or fetus when tenecteplase was given at doses up to 5 mg/kg daily during early gestation or as a single dose during mid gestation (approximately 8 times maximum clinical exposure, based on AUC).
Studies in animals have not been done to assess the effects of tenecteplase on general reproductive capacity or on offspring development after exposure in utero.
There are no adequate or well controlled studies in pregnant women. Tenecteplase should be given to pregnant women only if the potential benefits justify the potential risk to the fetus.
It is not known if tenecteplase is excreted into human milk. Because many drugs are excreted into human milk, caution should be exercised when Metalyse is administered to breastfeeding women and a decision must be made whether breast-feeding should be discontinued for the first 24 hours after administration of Metalyse. Studies in animals have not been done to assess the effect of tenecteplase on neonatal development. 4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at https://www.tga.gov.au/safety/reporting-problems.
Summary of the safety profile.
As with other thrombolytic agents, haemorrhage is the most common adverse reaction associated with the use of Metalyse. Haemorrhage at any site or body cavity can occur and may result in life-threatening situations, permanent disability or death.
The type of haemorrhage associated with thrombolytic therapy can be divided into two broad categories:
internal bleeding at any site or body cavity;
superficial bleeding, normally from injection sites.
With intracranial haemorrhage neurological symptoms such as somnolence, aphasia, hemiparesis, convulsion may be associated.
Serious adverse events (SAEs) reported in the Alteplase Compared to Tenecteplase (AcT) trial comparing the tenecteplase and alteplase treatment groups are provided in Table 2.
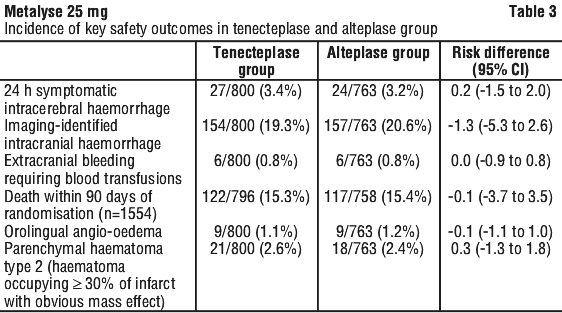
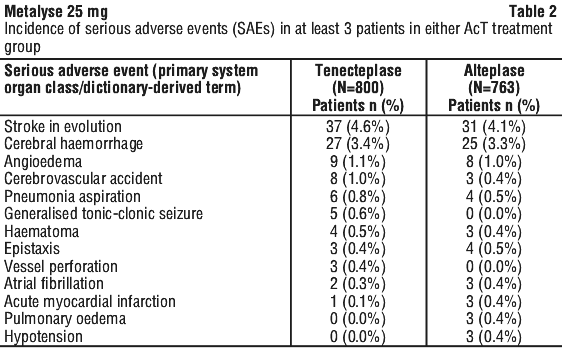 The most frequent undesirable effect of thrombolysis with tenecteplase is bleeding (haemorrhage) including symptomatic intracerebral haemorrhage in acute ischaemic stroke. In the AcT study the key safety outcomes were symptomatic intracerebral haemorrhage, extracranial bleeding requiring blood transfusion, and orolingual angio-oedema (all occurring within 24 h of thrombolytic administration), and 90-day all-cause mortality. A comparison of the tenecteplase and alteplase treatment groups is provided in Table 3.
The most frequent undesirable effect of thrombolysis with tenecteplase is bleeding (haemorrhage) including symptomatic intracerebral haemorrhage in acute ischaemic stroke. In the AcT study the key safety outcomes were symptomatic intracerebral haemorrhage, extracranial bleeding requiring blood transfusion, and orolingual angio-oedema (all occurring within 24 h of thrombolytic administration), and 90-day all-cause mortality. A comparison of the tenecteplase and alteplase treatment groups is provided in Table 3.
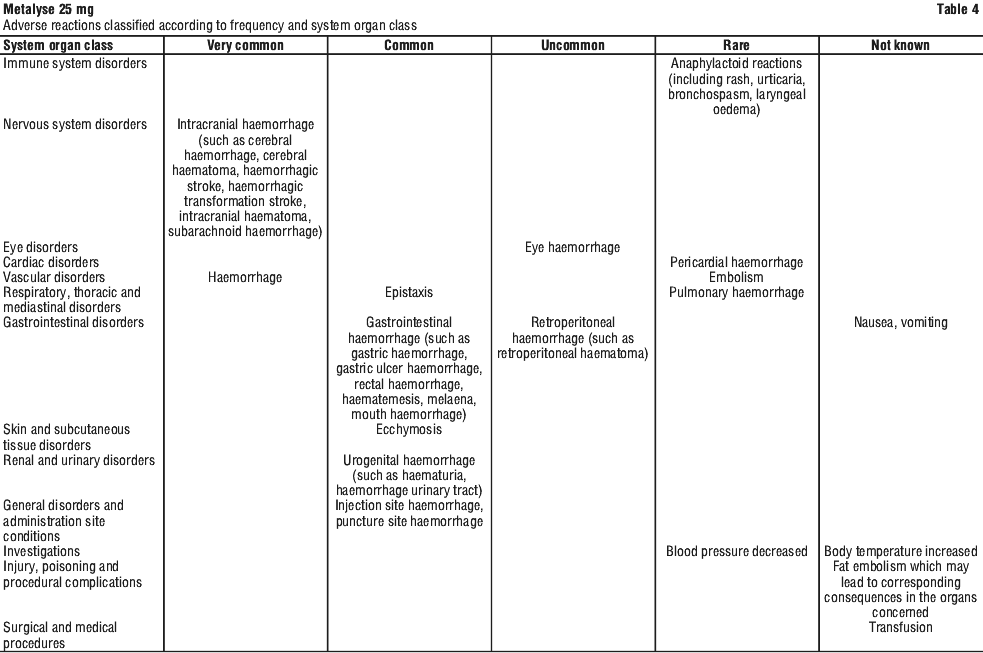
Tabulated list of adverse reactions.
The corresponding frequency category estimation for each adverse drug reaction is based on the following convention: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from the available data). See Table 4.
4.9 Overdose
For information on the management of overdose, contact the Poison Information Centre on 13 11 26 (Australia).
Symptoms.
In the event of overdose there may be an increased risk of bleeding.
Therapy.
In case of severe prolonged bleeding, substitution therapy may be considered (plasma, platelets) (also see Section 4.4 Special Warnings and Precautions for Use).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antithrombotic agents, enzymes.
ATC code: B01A D11.
Mechanism of action.
Tenecteplase is a tissue plasminogen activator (t-PA) produced by recombinant DNA technology, using an established mammalian cell line (Chinese Hamster Ovary cells).
Tenecteplase is a recombinant fibrin-specific plasminogen activator that is derived from native t-PA by modifications at three sites of the protein structure. It binds to the fibrin component of the thrombus (blood clot) and converts thrombus-bound plasminogen to plasmin, which degrades the fibrin matrix of the thrombus. Tenecteplase has a higher fibrin specificity and greater resistance to inactivation by its endogenous inhibitor compared to native t-PA.
After administration of tenecteplase, dose dependent consumption of α2-antiplasmin (the fluid-phase inhibitor of plasmin) with consequent increase in the level of systemic plasmin generation have been observed. This observation is consistent with the intended effect of plasminogen activation. In comparative studies, a less than 15% reduction in fibrinogen and a less than 25% reduction in plasminogen were observed in subjects treated with the maximum dose of tenecteplase (10,000 U, corresponding to 50 mg), whereas alteplase caused an approximately 50% decrease in fibrinogen and plasminogen levels. No clinically relevant antibody formation was detected at 30 days.
Clinical trials.
AcT study.
The Alteplase Compared to Tenecteplase (AcT) trial, was designed as a pragmatic, registry based, prospective, randomised, open label, controlled trial of intravenous tenecteplase vs. intravenous alteplase to provide evidence that tenecteplase is non-inferior to alteplase in patients with acute ischemic stroke within 4.5 h from last known well otherwise eligible for intravenous thrombolysis as per current guidelines. The trial achieved its primary outcome demonstrating a clinically relevant non inferiority with tenecteplase 0.25 mg/kg (max. 25 mg) vs alteplase 0.9 mg/kg (max. 90 mg): 296 (36.9%) of 802 patients in the tenecteplase group and 266 (34.8%) of 765 in the alteplase group had a modified Rankin Scale (mRS) score of 0-1 at 90-120 days (unadjusted risk difference 2.1% [95% CI - 2.6 to 6.9], meeting the prespecified non-inferiority threshold of -5%).
Key safety outcomes were symptomatic intracerebral haemorrhage, orolingual angio-oedema, and extracranial bleeding requiring blood transfusion, all occurring within 24 h of thrombolytic administration, and 90-day all-cause mortality.
There were no meaningful differences in the rate of 24 h symptomatic intracerebral haemorrhage. Rates of imaging-defined intracranial haemorrhage (assessed blinded to symptom status and treatment allocation) showed no differences between the two groups, and the imaging-defined rates of type 2 parenchymal haematoma (i.e. haematoma occupying ≥ 30% of infarct with obvious mass effect) were similar to the observed rates of symptomatic intracerebral haemorrhage in the trial. There were no meaningful differences in the rate of 90-day mortality 90 days from treatment. Orolingual angio-oedema and peripheral bleeding requiring blood transfusion were rare and similar in both groups (see Section 4.8 Adverse Effects (Undesirable effects), Table 3).
EXTEND-IA TNK study.
EXTEND-IA TNK was designed to assess whether tenecteplase is non-inferior to alteplase in achieving reperfusion at initial angiogram when administered within 4.5 h of ischaemic stroke onset in patients planned to undergo endovascular therapy.
Patients with ischaemic stroke who had occlusion of the internal carotid, basilar, or middle cerebral artery and who were eligible to undergo thrombectomy were randomised to receive tenecteplase 0.25 mg/kg or alteplase 0.9 mg/kg within 4.5 h after symptom onset. There were 101 patients in each treatment group. The primary outcome was reperfusion of greater than 50% of the involved ischaemic territory or an absence of retrievable thrombus at the time of the initial angiographic assessment. Non-inferiority of tenecteplase was tested, followed by superiority. Secondary outcomes included the mRS score at 90 days.
The primary outcome occurred in 22% of the patients treated with tenecteplase vs 10% of those treated with alteplase (incidence difference, 12%; 95% CI 2, 21; incidence ratio, 2.2; 95% CI 1.1, 4.4; p = 0.002 for non-inferiority; p = 0.03 for superiority).
Analysis of one of the several secondary outcomes, the mRS score at 90 days showed, that patients in the tenecteplase group had a median score of 2 (interquartile range, 0 to 3), which indicated significantly better function than the median score of 3 (interquartile range, 1 to 4) among patients in the alteplase group (common odds ratio, 1.7; 95% CI, 1.0 to 2.8; p = 0.04). There was no significant difference in the incidence of recovery to independent function (mRS score of 0 to 2 or no change from baseline function) at day 90, which occurred in 65 of 101 patients (64%) in the tenecteplase group and in 52 of 101 (51%) in the alteplase group (adjusted incidence ratio, 1.2; 95% CI, 1.0 to 1.5; p = 0.06; adjusted odds ratio, 1.8; 95% CI, 1.0 to 3.4; p = 0.06).
The proportion of mRS 0-1 at 90 days was 51% for the tenecteplase group and 43% for the alteplase group (p = 0.23).
The symptomatic intracerebral haemorrhage (sICH) occurred in 1% of the patients in each group. There were 10 deaths (10%) in the tenecteplase group and 18 (18%) in the alteplase group, which was not significant in the pre-specified logistic-regression analysis. Most of the deaths were related to progression of major stroke (9 in tenecteplase group and 14 in alteplase group). Tenecteplase 0.25 mg/kg showed a similar safety profile compared to alteplase 0.9 mg/kg.
Real world evidence.
Several non-interventional studies compared tenecteplase (0.25 mg/kg) versus alteplase (0.9 mg/kg) in AIS with or without large vessel occlusion (LVO) within 4.5 hours after symptom onset. These observational studies reported adjusted (or propensity score matched) estimates, included in total > 2,900 AIS patients (from studies with over 100 patients treated with tenecteplase), and reported a consistent similar or favourable safety and effectiveness profile of tenecteplase in comparison with intravenous alteplase. Endpoints measured included functional outcome (3-month mRS score), all-cause mortality, intracranial haemorrhage and symptomatic intracranial haemorrhage, rates of angioedema, door-to needle time, door-in-door out time, imaging-to-thrombolysis time, thrombolysis-to-puncture time, and onset-to-needle time.
5.2 Pharmacokinetic Properties
Absorption and distribution.
Tenecteplase is an intravenously administered, recombinant protein that activates plasminogen. Following i.v. bolus administration of 30 mg tenecteplase in patients with acute myocardial infarction, the initially estimated tenecteplase plasma concentration was 6.45 ± 3.60 microgram/mL (mean ± SD). The distribution phase represents 31% ± 22% to 69% ± 15% (mean ± SD) of the total AUC following the administration of doses ranges from 5 to 50 mg. The mean residence time (MRT) in the body is approximately 1 h and the mean (± SD) volume of distribution at the steady-state (Vss) ranged from 6.3 ± 2 L to 15 ± 7 L. A total of 113 AMI patients were enrolled and blood sampling for pharmacokinetics was conducted in 82 patients (72 male and 10 female).
Data on tissue distribution were obtained in studies with radioactively labelled tenecteplase in rats. The main organ to which tenecteplase distributed was the liver.
Metabolism.
Tenecteplase is cleared from the circulation by binding to specific receptors in the liver followed by catabolism to small peptides. Binding to hepatic receptors is, however, reduced compared to native t-PA, resulting in a prolonged half-life.
Excretion.
Extensive pharmacokinetic characterisation of tenecteplase was performed during Phase I and Phase II clinical trials. After single intravenous bolus injection of tenecteplase in patients with acute myocardial infarction, tenecteplase antigen exhibits biphasic elimination from plasma. There is no dose dependence of tenecteplase clearance in the therapeutic dose range. The initial, dominant half-life was 24 ± 5.5 (mean ± SD) min, which was 5 times longer than native t-PA. The terminal half-life was 129 ± 87 min, and plasma clearance was 119 ± 49 mL/min.
Increasing body weight resulted in a moderate increase of tenecteplase clearance, and increasing age resulted in a slight decrease of clearance. In general, women exhibit lower clearance than men, but this may be explained by the lower body weight of women.
Linearity/non-linearity.
The dose linearity analysis based on AUC suggested that tenecteplase exhibits non-linear pharmacokinetics in the dose range studied, i.e. 5 to 50 mg.
Special populations.
Renal and hepatic impairment.
As the kidneys do not appear to be involved in the elimination of tenecteplase, it is not expected that renal dysfunction will affect the pharmacokinetics. The effect of hepatic dysfunction on pharmacokinetics of tenecteplase in humans is not known.
5.3 Preclinical Safety Data
Genotoxicity.
Studies of tenecteplase in animals have not been performed to assess its mutagenicity.
Carcinogenicity.
Studies of tenecteplase in animals have not been performed to assess the carcinogenic potential.6 Pharmaceutical Particulars
6.1 List of Excipients
The excipients are arginine, phosphoric acid and polysorbate 20.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C. Before use, keep in the outer carton in order to protect from light.
The reconstituted solution has been demonstrated to be stable for 24 hours at 2-8°C or for 6 hours at room temperature.
To reduce microbiological hazard, the product should be used immediately after reconstitution. If storage of the reconstituted solution is necessary, it should be held at 2-8°C for not more than 24 hours and is the responsibility of the user.
6.5 Nature and Contents of Container
Metalyse 25 mg: 1 glass vial containing 25 mg (5,000 U) tenecteplase.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Tenecteplase is a 527 amino acid glycoprotein developed by introducing the following modifications to the complementary DNA (cDNA) for natural human t-PA: a substitution of threonine 103 with asparagine, and a substitution of asparagine 117 with glutamine, both within the kringle 1 domain, and a tetra-alanine substitution at amino acids 296-299 in the protease domain. Cell culture is carried out in nutrient medium containing the antibiotic gentamicin (65 mg/L). However, the presence of the antibiotic is not detectable in the final product (limit of detection is 0.67 microgram/vial).
CAS number.
191588-94-0.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes
