What is in this leaflet
This leaflet answers some common questions about Metalyse.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given Metalyse against the benefits they expect it will have for you.
If you have any concerns about being treated with Metalyse, ask your doctor or pharmacist.
This leaflet was last updated on the date at the end of this leaflet. More recent information may be available. The latest Consumer Medicine Information is available from your pharmacist, doctor, or from www.medicines.org.au and may contain important information about the medicine and its use of which you should be aware.
Keep this leaflet. You may need to read it again.
What Metalyse is used for
Metalyse is used during the early stages of a heart attack.
This medicine belongs to a group of medicines called tissue plasminogen activators (t-PA).
It works by dissolving clots in the blood vessels. These clots cause disease by interfering with normal blood flow.
Ask your doctor if you have any questions about why this medicine is being given to you. Your doctor may have prescribed it for another reason.
Before you are given Metalyse
When you must not be given it
You must not be given Metalyse if you have an allergy to:
- any medicine containing tenecteplase
- gentamicin (a trace residue from the manufacturing process)
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Metalyse must not be given to you if percutaneous coronary intervention (PCI) is planned.
PCI is an interventional procedure in which blocked blood vessels in the heart are unblocked. Examples of PCI procedures include balloon angioplasty, atherectomy and stent placement.
Receiving Metalyse treatment prior to a planned PCI procedure may increase your risk for side effects.
Because of the risk of bleeding, Metalyse must not be given to you if you have, or have had:
- current bleeding or severe bleeding in the past 6 months
- a family history of bleeding disorders
- treatment with an anti-clotting agent (anticoagulant), such as warfarin, unless its effect has had time to wear off
- major surgery, biopsy or significant trauma in the past 2 months
- a stroke due to bleeding in the brain or a stroke of unknown origin at any time
- a stroke caused by a blood clot or a transient ischaemic attack (TIA) in the past 6 months
- severe and uncontrolled high blood pressure (hypertension)
- tumours in which the risk of bleeding is increased
- any blood clotting defect
- current treatment with a thrombolytic agent (medicine used to dissolve blood clots)
- previous or current aneurysms (swelling and weakening of a part of a blood vessel) in your brain or spinal cord, or arteries in other parts of your body
- previous brain or spinal cord surgery
- previous or current tumours in your brain or spinal cord
- recent trauma to your head or skull
- heart and lung resuscitation (CPR) in the past 2 weeks
- structural abnormalities in your arteries or veins
- severe liver disease
- inflammation, infection or swelling of your heart or pancreas
- childbirth, organ biopsy or any invasive medical procedure in the past 10 days
- recent severe bleeding, particularly in your stomach, gut or from your genitals
- problems with your blood, especially if you also have severe liver or kidney disease
- stomach ulcers in the past 3 months.
Do not give this medicine to a child. Safety and effectiveness in children have not been established.
Metalyse must not be given after the expiry date printed on the pack or vial or if the packaging is torn or shows signs of tampering.
If you are not sure whether you should be given this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- any recent major trauma, medical procedure (such as a biopsy or injection) or surgery to any part of your body
- problems with blood supply to the brain
- a previous stroke caused by a blood clot or a transient ischaemic attack (TIA) more than 6 months previously
- recent bleeding from your stomach, gut or genitals
- high blood pressure
- any heart conditions or infections
- severe problems with your pancreas
- severe liver disease
- problems with your blood, especially if you also have severe liver or kidney disease
- diabetes mellitus
- bleeding from inside or around your eyes
- current treatment with an anti-clotting agent (anticoagulant), such as warfarin
- current treatment with a thrombolytic agent (medicine used to dissolve blood clots)
- if your body weight is less than 60 kg
- if you are elderly (75 years of age or older).
If you are uncertain as to whether you have, or have had, any of these conditions you should raise those concerns with your doctor.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell him/her before you are given Metalyse.
In addition, before starting treatment, your doctor will assess other factors which may increase the risks of using Metalyse.
These include infected veins and cannula sites or any condition in which bleeding is a significant risk or would be particularly difficult to manage because of its location.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Metalyse may interfere with each other. These include anticoagulants and platelet aggregation inhibitors, e.g. warfarin, low molecular weight heparin or any other medicines which affect the ability of the blood to clot.
These medicines may be affected by Metalyse, or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while being given this medicine.
How Metalyse is given
How much is given
The recommended dose is based on your body weight and ranges between 30 and 50 mg. The dose is given as a single injection over about 10 seconds.
Your doctor may prescribe a different dose or duration of treatment to that described here.
If you want more information, ask your doctor.
How it is given
Metalyse is a powder which must be mixed with sterile water for injections before being given into a vein through a drip line.
At the same time or soon after treatment with Metalyse, you may also receive other medications such as heparin to help prevent the blood vessel(s) becoming blocked again after treatment.
Metalyse should only be given under the supervision of a doctor and in a setting where appropriate equipment is readily available for diagnosis and patient monitoring.
You should only receive one injection of Metalyse. Any leftover solution that was prepared to treat you should be thrown away and not injected into anyone else.
When it is given
Treatment with Metalyse should begin as soon as possible after the onset of symptoms.
If you are given too much (overdose)
Overdose is unlikely because Metalyse is administered under medical supervision.
Symptoms of an overdose may include bleeding.
In the case of serious bleeding, your doctor will immediately stop treatment with Metalyse and heparin. Your doctor will start appropriate treatment to control the bleeding and, if necessary, replace the lost blood.
While you are being given Metalyse
Things you must do
Tell any other doctors, dentists, and pharmacists who treat you that you have been given this medicine.
Metalyse increases the risk of bleeding and bruising. After treatment with Metalyse medical staff will avoid giving you injections or moving you unless absolutely necessary.
Your doctor will probably continue to treat you with other medications after treatment with Metalyse. This is to reduce the risk of more blood clots forming.
Side effects
Tell your doctor or nurse as soon as possible if you do not feel well while you are being given Metalyse.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
If you are over 75 years of age you may have an increased chance of getting side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or nurse to answer any questions you may have.
The most common side effect is bleeding. This may have an effect on your blood readings.
Bleeding may be obvious if it is from the skin, nose or eyes. A more serious situation is when bleeding occurs inside the body (internally), for example, bruising and stroke (bleeding in the brain).
Other symptoms such as drowsiness, difficulty speaking, inability to move parts of your body and convulsion may also occur if you experience bleeding in the brain.
Internal bleeding can occur at any site or body cavity and may result in life-threatening situations, permanent disability or death.
Other side effects reported include nausea, vomiting, low blood pressure, irregular heart beat and fever. These events commonly occur after a heart attack and may or may not be caused by Metalyse.
There have also been reports of blockages of blood vessels following treatment with Metalyse. This can lead to organ failure (e.g. kidney failure). These serious effects are rare.
There have also been reports of serious or life-threatening allergic reactions, which can cause low blood pressure and difficulty breathing.
Tell your doctor as soon as possible if you experience any side effects during or after treatment with Metalyse, so that these may be properly treated.
Other side effects not listed above may also occur in some people.
Tell your doctor or nurse if you notice anything unusual, during or after treatment with Metalyse.
After being given Metalyse
Storage
Metalyse will be stored in the pharmacy or ward below 30°C and protected from light.
After mixing with sterile water for injections, Metalyse can be kept for up to 24 hours in a refrigerator (2-8°C).
Disposal
Each vial of Metalyse can only be used once and unused contents of opened vials must be discarded.
Product Description
What it looks like
Metalyse is the brand name of your medicine.
It comes as a sterile, white to off-white powder in clear glass vials. Metalyse powder must be mixed with sterile water for injections before use. When mixed, the resulting solution is colourless to pale yellow.
Metalyse is sold as a pack containing one vial of powder and one pre-filled syringe of sterile water for injections.
Ingredients
Each Metalyse 40 mg powder for injection vial contains 40 mg of tenecteplase. The pre-filled syringe contains 8 mL of sterile water for injections.
Each Metalyse 50 mg powder for injection vial contains 50 mg of tenecteplase. The pre-filled syringe contains 10 mL of sterile water for injections.
The reconstituted solution contains 5 mg tenecteplase (rch) per mL.
The powder for injection also contains:
- arginine
- phosphoric acid
- polysorbate 20.
Supplier
Metalyse is supplied in Australia by:
Boehringer Ingelheim Pty Limited
ABN 52 000 452 308
Sydney, Australia
www.boehringer-ingelheim.com.au
This Consumer Medicine Information was updated in July 2020.
® Metalyse is a registered trademark of Boehringer Ingelheim
© Boehringer Ingelheim Pty Limited 2020
Australian Registration Number
40 mg: AUST R 75012
50 mg: AUST R 75013
Published by MIMS September 2020

 Death and permanent disability have been reported in patients who have experienced stroke (including intracranial bleeding) and other serious bleeding episodes.
Death and permanent disability have been reported in patients who have experienced stroke (including intracranial bleeding) and other serious bleeding episodes. Nonintracranial major bleeding and the need for blood transfusions were lower in patients treated with Metalyse.
Nonintracranial major bleeding and the need for blood transfusions were lower in patients treated with Metalyse. As with other thrombolytic agents, the following events have been reported as sequelae of myocardial infarction and/or thrombolytic administration.
As with other thrombolytic agents, the following events have been reported as sequelae of myocardial infarction and/or thrombolytic administration. Rates of mortality and the combined endpoint of death or stroke among pre-specified subgroups, including age, gender, time to treatment, infarct location, and history of previous myocardial infarction, demonstrate consistent relative risks across these subgroups. There was insufficient enrollment of non-Caucasian patients to draw any conclusions regarding relative efficacy in racial subsets.
Rates of mortality and the combined endpoint of death or stroke among pre-specified subgroups, including age, gender, time to treatment, infarct location, and history of previous myocardial infarction, demonstrate consistent relative risks across these subgroups. There was insufficient enrollment of non-Caucasian patients to draw any conclusions regarding relative efficacy in racial subsets.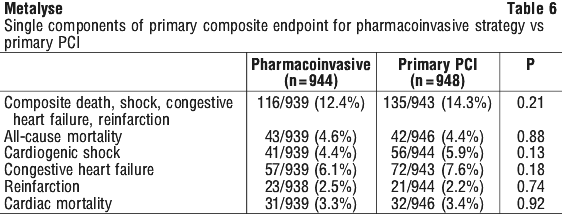 The observed incidence of major and of minor non-ICH bleeds were similar in both groups (Table 7).
The observed incidence of major and of minor non-ICH bleeds were similar in both groups (Table 7).
 None of the differences between groups displayed in Tables 7 and 8 reached the threshold of statistical significance except for the incidence of total strokes and ICH, however the incidences in the pharmacoinvasive group were as observed in previous clinical studies.
None of the differences between groups displayed in Tables 7 and 8 reached the threshold of statistical significance except for the incidence of total strokes and ICH, however the incidences in the pharmacoinvasive group were as observed in previous clinical studies.