

What is in this leaflet
This leaflet answers some common questions about Micardis.
It does not contain all available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of you taking this medicine against the benefits it is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
This leaflet was last updated on the date at the end of this leaflet. More recent information may be available. The latest Consumer Medicine Information is available from your pharmacist, doctor, or from www.medicines.org.au and may contain important information about the medicine and its use of which you should be aware.
Keep this leaflet with the medicine. You may need to read it again.
What Micardis is used for
Micardis is used to:
- treat high blood pressure (also called hypertension)
- prevent cardiovascular complications, including death due to cardiovascular causes, in patients older than 55 years of age with coronary artery disease, peripheral vascular disease, previous stroke, previous transient ischaemic attack (TIA) or high risk diabetes with evidence of end organ damage.
Treatment of Hypertension
Micardis is used to lower high blood pressure (hypertension).
Everyone has blood pressure. This pressure helps your blood move around your body. Your blood pressure may be different at different times of the day, depending on how busy or worried you are. You have hypertension (high blood pressure) when your blood pressure stays higher than normal, even when you are calm or relaxed.
There are usually no signs of hypertension. The only way of knowing that you have hypertension is to have your blood pressure checked on a regular basis. If high blood pressure is not treated, it can lead to serious health problems, including stroke, heart disease and kidney failure.
How Micardis works
Micardis contains telmisartan. Telmisartan belongs to a group of medicines called angiotensin II receptor blockers. Angiotensin II is a substance in the body which causes blood vessels to narrow, thus increasing blood pressure. Telmisartan works by blocking the effect of angiotensin II. When the effect of angiotensin II is blocked, your blood vessels relax and your blood pressure goes down.
Micardis may be used either alone or in combination with other medicines used to treat high blood pressure.
Prevention of Cardiovascular Complications, including Death due to Cardiovascular Causes
Micardis is also used to prevent cardiovascular complications, including death due to cardiovascular causes that may arise in high risk patients older than 55 years of age. Examples include heart attack, stroke, death caused by heart diseases or hospitalisation due to heart failure (a condition which can cause shortness of breath or ankle swelling).
Patients who may be considered at high risk of developing cardiovascular complications or at high risk of death due to cardiovascular causes are those aged 55 or more who have problems such as coronary artery disease (a heart disease caused by poor blood flow in the blood vessels of the heart), peripheral vascular disease (poor circulation in the hands or feet), previous stroke, previous transient ischaemic attack (TIA) or diabetes with additional high risk factors and evidence of end organ damage (e.g. damage occurring in the kidneys, heart, brain or eyes).
Your doctor can tell you if you are at high risk of developing cardiovascular complications or if you are at high risk of death due to cardiovascular causes.
Your doctor may have prescribed Micardis for another reason.
Ask your doctor if you have any questions about why Micardis has been prescribed for you.
Micardis is not addictive.
This medicine is available only with a doctor's prescription.
Before you take Micardis
When you must not take it
Do not take Micardis if you have an allergy to:
- any medicine containing telmisartan (the active ingredient in Micardis)
- any other ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take Micardis if you have a rare hereditary condition of fructose intolerance. The maximum recommended daily dose of Micardis contains approximately 338 mg of sorbitol.
Do not take Micardis if you are pregnant. Like other similar medicines, it may affect your developing baby if you take it during pregnancy.
Do not breastfeed if you are taking Micardis. It is not known if telmisartan, the active ingredient in Micardis, passes into breast milk and there is a possibility that your baby may be affected.
Do not give Micardis to a child under the age of 18 years. Safety and effectiveness in children and teenagers up to 18 years of age have not been established.
Do not take Micardis if you suffer from:
- severe liver disease
- biliary obstructive disorders (problem with the flow of bile from the gall bladder)
- diabetes or kidney problems and you are taking aliskiren (a medicine used to treat high blood pressure).
Do not take Micardis after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
Do not take Micardis if the tablets are discoloured.
If you are not sure whether you should start taking Micardis, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- kidney problems
- liver problems
- heart problems
- diabetes
- a condition known as primary hyperaldosteronism (raised aldosterone levels, also known as Conn's syndrome)
- fructose intolerance
- recent severe diarrhoea or vomiting.
Tell your doctor if you are following a very low salt diet.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell your doctor before you start taking Micardis.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Micardis may interfere with each other. These include:
- ramipril or any other medicines used to treat high blood pressure or heart problems
- potassium supplements or potassium- containing salt substitutes
- medicines or salt substitutes which may increase your potassium levels
- diuretics or fluid tablets, medicines used to help the kidneys get rid of salt and water by increasing the amount of urine produced
- nonsteroidal anti-inflammatory agents such as aspirin or ibuprofen (medicines used to relieve pain, swelling and other symptoms of inflammation including arthritis)
- lithium, a medicine used to treat certain mental illnesses
- digoxin, a medicine used to treat heart failure
- trimethoprim, a medicine used to treat bacterial infections
- heparin, a medicine used to thin your blood
- corticosteroids, medicines used to treat inflammatory conditions
- immunosuppressants, such as ciclosporin or tacrolimus (medicines used to prevent organ rejection after transplantation).
These medicines may be affected by Micardis, or may affect how well it works. Other medicines used to treat high blood pressure or medicines with blood pressure lowering potential may have an additive effect with Micardis in lowering your blood pressure. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking Micardis.
How to take Micardis
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
Your doctor or pharmacist will tell you how many tablets you will need to take each day. This depends on your condition and whether or not you are taking any other medicines.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
For the Treatment of Hypertension
The usual dose of Micardis for adults is one 40 mg tablet once a day.
If your blood pressure is still too high after 4-8 weeks of starting treatment, your doctor may increase your dose to 80 mg.
For the Prevention of Cardiovascular Complications, including Death due to Cardiovascular Causes
The usual dose of Micardis is one 80 mg tablet once a day.
Depending on how you respond to the treatment, your doctor may suggest a higher or lower dose.
It is important to take Micardis exactly as your doctor or pharmacist has told you.
How to take it
Swallow the tablet whole with a full glass of water.
When to take it
Take Micardis at about the same time each day, either morning or evening. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take Micardis before or after food.
How long to take it
Continue taking Micardis for as long as your doctor tells you. Micardis helps to control your high blood pressure, and/or prevents you from developing cardiovascular complications, but does not cure it. It is important to keep taking Micardis every day even if you feel well.
People who have high blood pressure often feel well and do not notice any signs of this problem.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take the dose as soon as you remember, and then go back to taking it as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting unwanted side effects.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26) for advice or go to Emergency at your nearest hospital, if you think that you or anyone else may have taken too much Micardis.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you take too much Micardis you may feel dizzy, light-headed or faint. Your heartbeat may be faster or slower than usual. You may experience rapid, shallow breathing or cold, clammy skin. This is because your blood pressure is too low.
While you are taking Micardis
Things you must do
If you are about to be started on any new medicine, tell your doctor and pharmacist that you are taking Micardis.
Tell any other doctors, dentists and pharmacists who treat you that you are taking Micardis.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking Micardis. Micardis may affect other medicines used during surgery.
If you become pregnant while taking Micardis, tell your doctor immediately.
If you feel that Micardis is not helping your condition, tell your doctor or pharmacist.
Tell your doctor if, for any reason, you have not used Micardis exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Things you must not do
Do not take Micardis to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not stop taking Micardis or lower the dosage without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how Micardis affects you. Like other medicines used to treat high blood pressure, Micardis may cause sleepiness, dizziness or lightheadedness in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
You may feel dizzy or light-headed when you begin to take Micardis, especially if you are also taking a diuretic (or fluid tablet) or if you are dehydrated.
If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly. Standing up slowly, especially when you get up from a bed or chair, will help your body get used to the change in position and blood pressure. If this problem continues or gets worse, talk to your doctor.
If you exercise, or if you sweat, or if the weather is hot, you should drink plenty of water.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Micardis.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Do not be alarmed by the following list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- dizziness or lightheadedness when you stand up especially when getting up from a sitting or lying position
- dizziness or spinning sensation, fainting
- tiredness or weakness
- 'flu-like' symptoms
- pain in the chest
- diarrhoea
- indigestion
- stomach pain or discomfort
- wind or excessive gas in the stomach or bowel (flatulence)
- upper respiratory tract infections
- shortness of breath
- back pain
- aching muscles, not caused by exercise (myalgia)
- muscle spasms or leg cramps or leg pain
- painful joints (arthralgia)
- tendon pain or tendonitis-like symptoms
- symptoms of urinary tract infections (including cystitis) such as burning sensation when passing urine, pain in the pelvis or mid-back, urine that appears cloudy, straining or pain when passing urine
- trouble sleeping (insomnia)
- feeling anxious
- depression
- fast or slow heart beats
- abnormal or blurred vision
- increased sweating
- dry mouth
- allergic skin reactions including skin rash (eczema); itchiness (pruritus); redness of the skin (erythema)
- symptoms that may indicate low blood sugar levels in the blood, such as sweating, weakness, hunger, dizziness, trembling, headache or numbness (especially in diabetic patients)
- abnormal liver functions
- symptoms that may indicate a worsening of the kidney function, such as passing little or no urine, drowsiness, nausea, vomiting, breathlessness, loss of appetite and weakness
- symptoms that may indicate high potassium levels in the blood, such as nausea, diarrhoea, muscle weakness and changes in heart rhythm
- symptoms that may indicate low sodium levels in the blood, such as headache, dizziness, confusion, forgetfulness, weakness, unsteadiness or difficulty concentrating
- signs of anaemia such as tiredness, being short of breath when exercising, dizziness and looking pale
- bleeding or bruising more easily than normal (thrombocytopenia)
- symptoms that may indicate an infection of the blood, such as high fever, chills, headache, confusion and rapid breathing
- changes in your red or white blood cell levels may occur (such changes are usually detected by a blood test).
If any of the following happen, tell your doctor immediately or go to Emergency at your nearest hospital:
- swelling of the face, lips, mouth, tongue or throat which may cause difficulty in swallowing or breathing
- severe and sudden onset of itchy or raised skin rash, hives or nettle rash.
These are serious side effects. You may need urgent medical attention or hospitalisation. These side effects are rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After taking Micardis
Storage
Keep the tablets in the blister strip until it is time to take them. The blister pack protects the tablets from light and moisture.
Keep Micardis in a cool dry place where the temperature stays below 30°C.
Do not store Micardis or any other medicine in the bathroom or near a sink. Do not leave it in the car or on a window sill. Heat and dampness can destroy some medicines.
Keep Micardis where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking Micardis or the expiry date has passed, ask your pharmacist what to do with any that is left over.
Product description
What it looks like
Micardis is the brand name of your medicine.
Micardis tablets are available in two strengths: 40 mg and 80 mg tablets.
Micardis 40 mg tablets are white to off-white oblong tablets, marked with the Boehringer Ingelheim logo on one face and '51H' on the other.
Micardis 80 mg tablets are white to off-white oblong tablets, marked with the Boehringer Ingelheim logo on one face and '52H' on the other.
Micardis tablets are available in blister packs of 7 (sample), 28, 56* and 98* tablets.
* Not currently distributed in Australia.
Ingredients
Each Micardis 40 mg tablet contains 40 mg telmisartan.
Each Micardis 80 mg tablet contains 80 mg telmisartan.
The other ingredients found in both strengths are:
- povidone
- meglumine
- sodium hydroxide
- sorbitol
- magnesium stearate.
Supplier
Micardis tablets are supplied in Australia by:
Boehringer Ingelheim Pty Limited
ABN 52 000 452 308
Sydney, Australia
www.boehringer-ingelheim.com.au
® Micardis is a registered trademark of Boehringer Ingelheim
© Boehringer Ingelheim Pty Limited 2022
This Consumer Medicine Information was updated in July 2022.
Australian Registration Numbers
Micardis 40 mg tablets:
AUST R 68052
Micardis 80 mg tablets:
AUST R 68053
Published by MIMS September 2022
 In addition, the following adverse events occurred in more than 1% of the 3455 patients treated in all trials with telmisartan although causal association of these events with telmisartan could not be established: bronchitis, insomnia, arthralgia, anxiety, depression, palpitation, muscle spasms (cramps in legs) and rash.
In addition, the following adverse events occurred in more than 1% of the 3455 patients treated in all trials with telmisartan although causal association of these events with telmisartan could not be established: bronchitis, insomnia, arthralgia, anxiety, depression, palpitation, muscle spasms (cramps in legs) and rash. Combining telmisartan with ramipril in the ONTARGET study resulted in a higher incidence of hyperkalemia, renal failure, hypotension and syncope compared to telmisartan or ramipril alone.
Combining telmisartan with ramipril in the ONTARGET study resulted in a higher incidence of hyperkalemia, renal failure, hypotension and syncope compared to telmisartan or ramipril alone.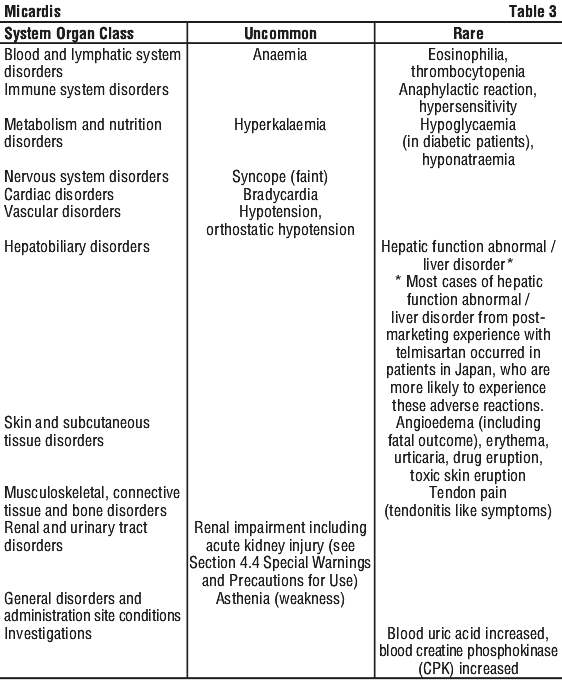
 Telmisartan is a specific angiotensin II receptor (type AT1) blocker. The chemical name for telmisartan is 4'-[(1,4'-dimethyl-2'-propyl [2,6'-bi-1H-benzimidazol]-1'-yl)- methyl]-[1,1'-biphenyl]- 2-carboxylic acid (IUPAC nomenclature). The molecular formula is C33H30N4O2 and the molecular weight is 514.6.
Telmisartan is a specific angiotensin II receptor (type AT1) blocker. The chemical name for telmisartan is 4'-[(1,4'-dimethyl-2'-propyl [2,6'-bi-1H-benzimidazol]-1'-yl)- methyl]-[1,1'-biphenyl]- 2-carboxylic acid (IUPAC nomenclature). The molecular formula is C33H30N4O2 and the molecular weight is 514.6.