1 Name of Medicine
Moxonidine.
2 Qualitative and Quantitative Composition
Moxonidine is a white to almost white powder with a melting point of 197-205°C. Moxonidine is very slightly soluble in water, sparingly soluble in methanol.
200 microgram.
Each tablet contains 200 microgram moxonidine.
400 microgram.
Each tablet contains 400 microgram moxonidine.
List of excipients with known effect.
Contains sugars as lactose.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Tablet, film coated.
Moxonidine-WGR 200 microgram tablets.
Pale pink, coloured round shaped tablet debossed with MX2 on one side and plain on another side.
Moxonidine-WGR 400 microgram tablets.
Dull brick red coloured round shaped tablets debossed with MX4 on one side and plan on another side.4.1 Therapeutic Indications
Moxonidine-WGR is indicated for the treatment of hypertension.
4.2 Dose and Method of Administration
Treatment should be started with 200 microgram Moxonidine-WGR in the morning. The dose may be titrated after two weeks to 400 microgram given as one dose or as divided doses (morning and evening) until a satisfactory response is achieved. If the response is still unsatisfactory after a further 2 weeks treatment, the dosage can be increased up to a maximum of 600 microgram in divided doses (morning and evening).
A single daily dose of 400 microgram and a divided daily dose of 600 microgram of Moxonidine-WGR should not be exceeded.
Moxonidine-WGR may be taken with or without food.
4.3 Contraindications
Hypersensitivity to any of the ingredients of this product.
Heart failure (NYHA Class I - IV).
Patients aged 75 years or older.
Bradycardia (HR < 50 beats/minute) or severe bradyarrhythmia, including sick sinus syndrome, or second or third degree atrioventricular (AV) block.
Malignant arrhythmias.
Severe renal impairment (GFR < 30 mL/min, serum creatinine concentration > 160 micromol/L).
4.4 Special Warnings and Precautions for Use
Renal impairment.
In patients with moderate renal impairment (GFR 30 - 60 mL/min), the single dose should not exceed 200 microgram and the daily dose should not exceed 400 microgram of moxonidine.
Use in the elderly.
See Section 4.3 Contraindications; Section 5.2 Pharmacokinetic Properties, Pharmacokinetics in the elderly.
Paediatric use.
Moxonidine-WGR should not be given to children below the age of 16 years as insufficient therapeutic experience exists in this group.
Effects on laboratory tests.
No data available.
Cessation of combination therapy with beta-blockers.
Abrupt cessation of combination therapy with Moxonidine-WGR and a beta-blocker may result in rebound hypertension. If combination therapy with Moxonidine-WGR and a beta-blocker is to be ceased, the beta-blocker should be stopped first and then Moxonidine-WGR stopped after a few days have elapsed. During cessation of therapy blood pressure should be regularly monitored.
Angioneurotic oedema.
As with other centrally acting antihypertensives, Moxonidine-WGR should be used with caution in patients with a history of angioneurotic oedema, severe coronary artery disease and unstable angina.
Cases of varying degrees of A-V block have been reported in the post-marketing setting in patients undergoing moxonidine treatment. Based on these case reports, the causative role of moxonidine in delaying atrioventricular conduction cannot be completely ruled out. Therefore, caution is recommended when treating patients with a possible predisposition to developing an A-V block.
When moxonidine is used in patients with 1st degree A-V block, special care should be exercised to avoid bradycardia. Moxonidine must not be used in higher degree A-V blocks (see Section 4.3 Contraindications).
In patients with moderate renal impairment (GFR > 30 - < 60 mL/min, serum creatinine > 105 - < 160 micromol/L), the hypotensive effect of Moxonidine-WGR should be closely monitored, especially at the start of treatment (see Section 4.2 Dose and Method of Administration).
Moxonidine-WGR should not be used because of lack of experience in cases of intermittent claudication, Raynaud's disease, Parkinson's disease, epileptic disorders, glaucoma, and depression.
Due to lack of therapeutic experience, the use of Moxonidine-WGR concomitantly with alcohol or tricyclic antidepressants should be avoided.
In limited studies, no rebound effect of the blood pressure after sudden discontinuation of moxonidine treatment has been detected. Nevertheless, it is advised not to interrupt the intake of Moxonidine-WGR abruptly. Moxonidine-WGR should be withdrawn gradually over a period of days.
Due to the presence of lactose in Moxonidine-WGR tablets, patients with rare hereditary problems of galactose intolerance, Lapp lactase deficiency, or glucose-galactose malabsorption should not take this medicine.4.5 Interactions with Other Medicines and Other Forms of Interactions
Concurrent administration of other antihypertensive agents enhances the hypotensive effect of Moxonidine-WGR.
The effect of sedatives and hypnotics may be intensified by Moxonidine-WGR. The sedative effect of benzodiazepines can be enhanced by concurrent administration of Moxonidine-WGR. Moxonidine moderately augmented the impaired performance in cognitive functions in subjects receiving lorazepam.
Since tricyclic antidepressants may reduce the effectiveness of centrally acting antihypertensive agents, it is not recommended that tricyclic antidepressants be co-administered with moxonidine. In healthy volunteers, no pharmacokinetic interactions have been observed with glibenclamide or digoxin.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Moxonidine did not affect fertility of rats at oral doses up to 6.4 mg/kg/day (15-64 times the clinical exposure at 0.3 mg BID, based on plasma moxonidine concentration).
(Category B3)
Moxonidine was not teratogenic in rats. It caused abortions, increased embryofoetal losses, and/or delayed foetal development at high oral doses (≥ 9 mg/kg/day in rats and 4.9 mg/kg/day in rabbits), associated with maternal toxicity. No adverse effects on embryofoetal development were seen at 2 mg/kg/day in rats and 0.6 mg/kg/day in rabbits. Exposures at the no effect dose were 24 (rat, based on AUC) and 22 (rabbit, based on dose adjusted for body surface area) times the clinical exposure at the maximum recommended clinical dose (0.3 mg BID).
There is inadequate data in pregnant women or women of childbearing age, and as such moxonidine should not be used during pregnancy unless the benefit clearly justifies the possible risk to the foetus.
Oral administration of moxonidine to rats from late pregnancy until weaning was associated with maternotoxicity, reduced pup weight and viability, and delayed pup development at ≥ 3 mg/kg/day, with no effects at 1 mg/kg/day (approximately 12 times the clinical exposure at 0.3 mg BID, based on AUC).
Studies in humans have shown that moxonidine passed from the maternal blood stream to breast milk. As the effect on the newborn infant is unknown, moxonidine should not be used by nursing mothers, unless the benefits clearly justify the possible risks to the infants.4.7 Effects on Ability to Drive and Use Machines
Based on its pharmacodynamic properties, moxonidine is unlikely to affect the ability to drive or operate machinery. However, it should be taken into account that occasionally dizziness or weariness may occur during treatment of hypertension.
4.8 Adverse Effects (Undesirable Effects)
Moxonidine has been evaluated for safety in over 4000 subjects worldwide, including more than 1200 patients treated for more than 6 months and over 800 patients treated for 1 year or longer. Most adverse events were of mild or moderate severity and did not require discontinuation of therapy.
In placebo-controlled clinical trials, 4.7% of patients treated with moxonidine discontinued therapy due to clinical adverse experiences, compared to 2.5% discontinuations among placebo-treated patients. Table 1 lists adverse events that occurred at an incidence of 1% or more among moxonidine-treated patients who participated in placebo-controlled trials of 6 to 8 weeks' duration, using doses of 200 microgram to 0.8 mg daily.

Laboratory test finding.
In controlled clinical trials, clinically important changes in standard laboratory parameters possibly associated with administration of moxonidine were rarely observed and occurred at rates comparable to those seen with placebo.
Post-marketing adverse events.
Common (≥ 1/100 and < 1/10): dry mouth, headache, dizziness, nausea, sleep disturbance, somnolence, asthenia, vasodilatation, anxiety.
Uncommon (≥ 1/1000 and < 1/100): sedation, insomnia, skin rash, urticaria, pruritus.
Rare (≥ 1/10,000 and < 1/1000): hypotension, postural hypotension.
Very rare (<1/10,000): angioedema.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Oral dosages up to 2.0 mg/day have been tolerated without the occurrence of serious adverse events. The following case of accidental overdose with moxonidine by a 2 year old child has been reported: the child ingested an unknown quantity of moxonidine. The maximum dosage possibly ingested was 14 mg. The child had the following symptoms: sedation, coma, hypotension, miosis and dyspnoea. Gastric lavage, glucose infusion, mechanically assisted ventilation and rest resulted in the complete disappearance of the symptoms in 11 hours.
Because of the pharmacodynamic properties of moxonidine, the following symptoms can be expected in adults: sedation, hypotension, orthostatic dysregulation, bradycardia, dry mouth. In rare cases emesis and paradoxical hypertension may occur. No specific treatment is known. Phentolamine (Regitine) may, depending on the dose, reverse part of the symptoms of moxonidine overdose. Measures to support blood circulation are recommended.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
In different animal models, moxonidine has been shown to be a relatively potent antihypertensive agent. Available experimental data suggest that the site of the antihypertensive action of moxonidine is the central nervous system (CNS). Moxonidine has been shown to bind to I1-imidazoline receptors and to a lesser extent α2-adrenoreceptors. The therapeutic action of moxonidine appears to result from interaction with I1 and α2-receptors located within the rostral ventrolateral medulla, leading to a reduced activity of sympathetic nerves.
Moxonidine differs from other available centrally acting antihypertensives by exhibiting only low affinity to central α2-adrenoceptors compared to I1-imidazoline receptors; α2-adrenoceptors are considered the molecular target via which sedation and dry mouth, the most common side effects of centrally acting antihypertensives, are mediated. In humans, moxonidine leads to a reduction of systemic vascular resistance and consequently in arterial blood pressure.
Clinical trials.
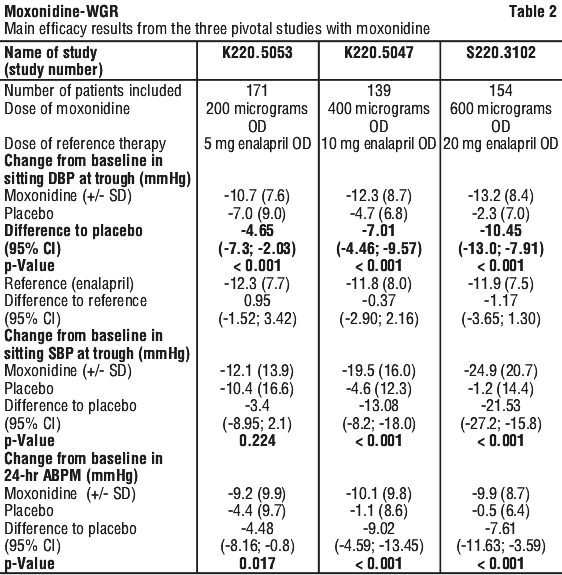
The antihypertensive effects of moxonidine were demonstrated in three placebo- and reference-controlled pivotal trials, studying dosages of 0.2 to 0.6 mg once daily in patients with mild to moderate hypertension (office diastolic blood pressure 95 - 110 mmHg; 24 hour ambulatory diastolic blood pressure ≥ 85 mmHg). The studies allowed direct comparison of moxonidine to a reference agent (enalapril) at equipotent doses, analysis of peak and trough effects, and (in pooled data) an evaluation of the dose response relationship. In all, 464 patients were included, of whom 152 were randomized to moxonidine. All studies were double-blind, placebo and reference-controlled, prospectively randomized, parallel group comparisons, with a 4 week placebo run-in period followed by 8 weeks double-blind treatment.
The first study compared 200 microgram moxonidine with 5 mg enalapril and placebo once daily. The intent-to-treat patient population comprised 169 patients evaluable for office blood pressure and 152 for ABPM. The mean placebo-subtracted differences in office systolic/diastolic blood pressure at trough from baseline to week 8 were -3.40/-4.65 mmHg for moxonidine. The reduction for diastolic blood pressure was statistically significant versus placebo (p < 0.001) and not different from enalapril. The difference in systolic blood pressure did not differ statistically from placebo. The overall incidence of adverse events of moxonidine (27.8%) was similar to enalapril (26.7%) and placebo (22.8%) in this trial.
In the second study of similar design, 400 microgram moxonidine was compared with 10 mg enalapril and placebo once daily. A total of 139 patients were included in the intent-to-treat sample for office blood pressure and 108 patients for ABPM. Moxonidine reduced office systolic and diastolic blood pressures at trough highly statistically significantly by 13.08/7.01 mmHg (placebo-corrected; p < 0.001 for both) and was equivalent to enalapril. Similar results were obtained with the 24 hour, daytime and night-time ABPM values. The 24 hour effects were maintained over the dosing interval with a trough to peak ratio of approximately 70%. Treatment emergent adverse events were reported in 36.6% of the moxonidine patients, in 31.9% of the enalapril patients and in 28.9% with placebo.
In the third pivotal study of similar design, 0.6 mg moxonidine was compared with 20 mg enalapril and placebo once daily. The intent-to-treat population consisted of 154 patients for office blood pressure measurements and 130 for ABPM. Office systolic and diastolic blood pressures were markedly and statistically significantly reduced by moxonidine by 21.53/10.45 mmHg (placebo-corrected; p < 0.001 for both) in comparison to placebo and did not differ from enalapril. Mean 24-hour ABPM was statistically reduced versus placebo for both diastolic and systolic blood pressures as well. Adverse events were recorded for 43.1% of patients in the moxonidine group in comparison to 32.1% in the enalapril group and 22.6% with placebo. The main efficacy results for the primary and secondary parameters can be taken from the Table 2.
 The combination of hydrochlorothiazide and moxonidine was evaluated in a further placebo-controlled, double-blind and prospectively randomized trial. The study comprised a total of 161 randomized patients: 37 received moxonidine 400 microgram, 40 received hydrochlorothiazide 25 mg, 42 received the combination 0.4/25 mg and 41 received placebo, all once daily. Sitting systolic and diastolic blood pressures at trough after eight weeks treatment were statistically significantly reduced for all active treatment arms versus placebo. Furthermore, the difference of the combination for sitting diastolic blood pressure was statistically significant in favour of the combination for both monotherapies. The incidence of treatment emergent adverse events did not differ significantly between groups: 32.5% with moxonidine, 25% with HCT, 30% with the combination and 23% with placebo. See Table 3.
The combination of hydrochlorothiazide and moxonidine was evaluated in a further placebo-controlled, double-blind and prospectively randomized trial. The study comprised a total of 161 randomized patients: 37 received moxonidine 400 microgram, 40 received hydrochlorothiazide 25 mg, 42 received the combination 0.4/25 mg and 41 received placebo, all once daily. Sitting systolic and diastolic blood pressures at trough after eight weeks treatment were statistically significantly reduced for all active treatment arms versus placebo. Furthermore, the difference of the combination for sitting diastolic blood pressure was statistically significant in favour of the combination for both monotherapies. The incidence of treatment emergent adverse events did not differ significantly between groups: 32.5% with moxonidine, 25% with HCT, 30% with the combination and 23% with placebo. See Table 3.

5.2 Pharmacokinetic Properties
Absorption.
In humans, about 90% of an oral dose of moxonidine is absorbed; it is not subject to first-pass metabolism and its bioavailability is 88%. Food intake does not interfere with moxonidine pharmacokinetics. The maximum plasma levels of moxonidine are reached 30 - 180 minutes after the intake of a film-coated tablet.
Distribution.
Only about 10% of moxonidine is bound to plasma proteins (Vdss=1.8 ± 0.4 L/kg).
Metabolism.
Moxonidine is 10 - 20% metabolised, mainly to 4,5-dehydromoxonidine and to a guanidine derivative by opening of the imidazoline ring. The hypotensive effect of 4,5-dehydromoxonidine is only 1/10, and that of the guanidine derivative is less than 1/100 of that of moxonidine.
Excretion.
Moxonidine and its metabolites are eliminated almost entirely via the kidneys. More than 90% of the dose is eliminated via the kidneys in the first 24 hours after administration, while only about 1% is eliminated via the faeces. The cumulative renal excretion of unchanged moxonidine is about 50 - 75%.
The mean plasma elimination half-life of moxonidine is 2.2 - 2.3 hours, and renal elimination half-life is 2.6 - 2.8 hours. Although moxonidine has a relatively short half-life it should be administered no more frequently than twice daily.
Pharmacokinetics in the elderly.
In older patients, the exposure (AUC) to moxonidine increased by approximately 50% following a single dose and at steady state, therefore lower doses of moxonidine should be used for the treatment of hypertension (see Section 4.3 Contraindications).
Pharmacokinetics in renal impairment.
In moderately impaired renal function (GFR 30 - 60 mL/min), AUC increases by 85% and clearance decreased to 52%. In such patients the hypotensive effect of moxonidine should be closely monitored, especially at the start of treatment (see Section 4.2 Dose and Method of Administration).
5.3 Preclinical Safety Data
Genotoxicity.
Moxonidine did not induce gene mutations in bacteria or mammalian cells in vitro. Nor did it induce chromosomal aberrations in human lymphocytes in vitro or in Chinese hamster bone marrow cells in vivo.
Carcinogenicity.
Carcinogenicity studies in rats and mice at oral doses of up to 3.6 mg/kg (rat) and 7.4 mg/kg (mouse) did not reveal evidence of carcinogenic potential. Systematic exposures at the highest dose were approximately 3 (rats, based on plasma moxonidine concentration) and 63 (mice, based on and dose adjusted for body surface area) times the clinical exposure at 0.3 mg BID.6 Pharmaceutical Particulars
6.1 List of Excipients
Each tablet includes the following excipients: lactose monohydrate, crospovidone, povidone, magnesium stearate, Opadry complete film coating system 03F540200 Pink (PI 111786) for 200 microgram strength and Opadry complete film coating system 03F540201 Pink (PI 111784) for 400 microgram strength.
6.2 Incompatibilities
Concurrent administration of other antihypertensive agents enhances the hypotensive effect of Moxonidine-WGR.
The effect of sedatives and hypnotics may be intensified by Moxonidine-WGR. The sedative effect of benzodiazepines can be enhanced by concurrent administration of Moxonidine-WGR. Moxonidine moderately augmented the impaired performance in cognitive functions in subjects receiving lorazepam.
Since tricyclic antidepressants may reduce the effectiveness of centrally acting antihypertensive agents, it is not recommended that tricyclic antidepressants be co-administered with moxonidine.
In healthy volunteers, no pharmacokinetic interactions have been observed with glibenclamide or digoxin.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Moxonidine-WGR tablets should be stored below 25°C. Protect from light. Protect from moisture.
6.5 Nature and Contents of Container
Supplied in blister pack as follows:
Moxonidine-WGR 200 microgram tablets.
PVC/PVDC/Al blister pack of 7 tablets (sample), 10 tablets, 14 tablets, 28 tablets, 30 tablets, 56 tablets, 84 tablets and 98 tablets.
Moxonidine-WGR 400 microgram tablets.
PVC/PVDC/Al blister pack of 7 tablets (sample), 10 tablets, 14 tablets, 28 tablets, 30 tablets, 56 tablets, 84 tablets and 98 tablets.
Not all pack sizes will be marketed in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
Chemical name: 4-chloro-N-(imidazolidin-2-ylidine)-6-methoxy-2-methyl-5-pyrimidinamine. C9H12ClN5O, MW: 241.7.

CAS number.
75438-57-2.7 Medicine Schedule (Poisons Standard)
Schedule 4: Prescription Only Medicine.
Summary Table of Changes


