What is in this leaflet
This leaflet answers some common questions about MVASI infusion.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking MVASI against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What MVASI is used for
MVASI contains the active ingredient bevacizumab.
MVASI is a biosimilar medicine to AVASTIN®. The evidence for comparability supports the use of MVASI for the treatment of:
- brain tumours resistant to previous treatments
- metastatic (spreading) cancer of the large bowel (i.e. in the colon or rectum), breast or cervix in combination with chemotherapy agents
- lung cancer and cancer of the ovaries and fallopian tubes (which can extend to the lining of surrounding organs such as stomach, liver) in combination with chemotherapy agents
- kidney cancer (renal cell cancer) in combination with interferon therapy (Roferon-A®).
MVASI belongs to a group of medicines known as anti-neoplastic (or anti-cancer) agents. There are many different classes of anti-neoplastic agents. MVASI belongs to a class known as anti-angiogenic agents.
Anti-angiogenic agents inhibit angiogenesis (the process of forming new blood vessels in your body).
MVASI selectively binds to vascular endothelial growth factor (VEGF), a protein found on the cells that line blood vessels. Tumours produce high levels of VEGF, which stimulates blood vessels to grow, thereby providing the tumour with nutrients and oxygen.
When MVASI blocks VEGF it disrupts the blood supply to the tumour, stopping or slowing down its growth.
There are many different types of medicines used to treat brain tumours and metastatic cancer of the large bowel, breast, lung, kidney and cervix.
Your doctor may have prescribed MVASI for another purpose.
Ask your doctor if you have any questions about why MVASI has been prescribed for you.
MVASI is not addictive.
This medicine is available only with a doctor's prescription.
Before you are given
MVASI When you must not be given it
Do not use MVASI if:
- you have had an allergic reaction to MVASI or any ingredients listed at the end of this leaflet
Symptoms of an allergic reaction may include shortness of breath; wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body or rash, itching or hives on the skin
- you have had an allergic reaction to any proteins that are of Chinese hamster origin or to other recombinant human or humanised antibodies
- the package is torn or shows signs of tampering
- the expiry date (EXP) printed on the pack has passed.
If you take this medicine after the expiry date has passed, it may not work as well.
If you are not sure if you should be given MVASI, talk to your doctor.
Do not give MVASI to children and adolescents. Safety and effectiveness in children and adolescents have not been established.
Before you are given it
Tell your doctor if:
- you are pregnant or plan to become pregnant
Do not use MVASI if you are pregnant. MVASI may cause damage to your unborn baby.
You should use contraception during treatment with MVASI and for at least 6 months after your last dose. If you become pregnant while you are being treated with MVASI, immediately inform your doctor.
Your doctor will advise you about using contraception during treatment with MVASI.
- you plan to start a family in the future
MVASI may interfere with your ability to become pregnant. Your doctor will advise you of your options prior to starting treatment.
- you are breast-feeding or plan to breast-feed
You should not breast-feed while being treated with MVASI and for at least 6 months after the last dose. MVASI may interfere with the growth and development of your baby.
- you have any other health problems, especially the following:
- inflammation of the bowel (symptoms may include fever, vomiting, diarrhoea and stomach pain) or stomach ulcers,
- hypertension (high blood pressure) - it is important to follow all your doctor's instructions to control your blood pressure
- history of blood clots or stroke, or you are taking medicine to prevent blood clots (e.g. warfarin)
- you or anyone in your family suffer from bleeding problems
- heart disease
- history of diabetes
- you have had major surgery within the last 28 days or have a wound that has not healed properly
MVASI can cause an increased risk of post-operative bleeding or problems with wound healing.
- you have had a blocked lung artery (pulmonary embolism)
MVASI may increase the risk of recurrence
- you have ever received anthracyclines (e.g. doxorubicin), a specific type of chemotherapy used to treat some cancers or have had radiotherapy to your chest MVASI can increase the risk of developing a weak heart.
- if you have or have had pain in the mouth, teeth and/or jaw, swelling or sores inside the mouth, numbness or a feeling of heaviness in the jaw, or loosening of a tooth tell your doctor immediately.
You may be advised to have a dental check-up before you start treatment with MVASI.
- you are 65 years of age or older
MVASI can increase the risk of blood clots which can lead to strokes or heart attacks in patients older than 65 years of age compared with younger patients. MVASI can also increase the risk of fatigue, hair loss, reduce the number of white cells in the blood and cells which help blood clot, inflammation of the mouth or throat, high blood pressure and a feeling of numbness or tingling in the hands or feet in patients older than 65 years of age compared with younger patients.
- you are allergic to any other medicines, foods, dyes or preservatives
If you have not told your doctor about any of the above, tell them before you start taking MVASI.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you have bought from a pharmacy, supermarket or health food shop.
Tell your doctor if you have recently received, or are receiving, radiotherapy.
Tell your doctor if you have recently received, or are receiving, a bisphosphonate (for example medicines containing ibandronate sodium, zoledronic acid or disodium pamidronate).
Some medicines may interfere with MVASI.
Some medicines may be affected by MVASI or may affect how well it works. You may need to use different amounts of your medicine, or you may need to take different medicines. Your doctor will advise you.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking MVASI.
Ask your doctor or pharmacist if you are not sure about this list of medicines.
How MVASI is given
How it is given
MVASI solution is prepared by a health care professional.
MVASI is given by infusion into a vein (intravenous infusion) by a health care professional.
The first infusion is usually given over 90 minutes. If it is well tolerated the second infusion may be given over 60 minutes. Later infusions may be given over 30 minutes.
How much is given
Your dose depends on your body weight and the type of cancer to be treated. MVASI can be given either once every 2 weeks or once every 3 weeks. Your doctor will prescribe a dose of MVASI that is right for you.
If you have been given too much MVASI you may develop a severe migraine. If this happens tell your health care professional immediately.
How long is it given
The number of infusions you will receive depends on how you are responding to treatment. Your doctor will discuss this with you.
If you miss a dose
Your doctor will decide when you should be given your next dose of MVASI.
While you are being treated with MVASI
Things you must do
Tell all doctors, dentists and pharmacists who are treating you that you are being treated with MVASI.
Tell your doctor immediately if you become pregnant during treatment with MVASI, or plan to start a family in the near future.
Tell your doctor immediately if you are breast-feeding while being treated with MVASI.
Tell your doctor if you are planning to have surgery or you have a wound that is not healing properly.
Tell your doctor if you need to undergo an invasive dental treatment or dental surgery, in particular when you are also receiving or have received a bisphosphonate (for example medicines containing ibandronate sodium, zoledronic acid or disodium pamidronate)
Tell your doctor if you feel MVASI is not helping your condition.
Be sure to keep all of your appointments with your doctor so that your progress can be checked.
Things you must not do
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how MVASI affects you. MVASI has not been shown to impair your ability to drive or operate machinery.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while being treated with MVASI.
MVASI helps most people with brain tumours and cancer of the large bowel (i.e. colon or rectum), breast, lung, kidney, ovary/fallopian tube (which can extend to the lining of surrounding organs such as stomach, liver) and cervix but it may have unwanted side effects in some people.
All medicines can have some unwanted side effects. Sometimes they are serious, but most of the time they are not. Your doctor has weighed the risks of you being treated with MVASI against the benefits they expect it will have for you.
Because MVASI is used with other medicines that treat cancer (including chemotherapy), it may be difficult for your doctor to tell whether the side effects are due to MVASI or due to other medicines.
MVASI may exacerbate some chemotherapy side effects when used in combination with chemotherapy agents including hair loss, nail disorders, pain, redness and/or swelling of your hands and/or soles of your feet, and a feeling of numbness or tingling in the hands or feet.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Tell your doctor if you notice any of the following and they worry you:
- high blood pressure
- body pain
- muscle and joint pain
- lack of energy or tiredness
- diarrhoea; constipation or rectal bleeding
- inflammation of the mouth or swollen/stiff neck
- sore mouth; mouth ulcers; cold sores
- loss of appetite
- shortness of breath
- nose bleed; runny or blocked nose
- dry skin; rash; flaking, swelling or redness of the skin or change in skin colour
- pain, redness and/or swelling of your hands and/or the soles of your feet that has affected your normal activities (hand-foot syndrome)
- numbness or weakness of the arms and legs
- change in sense of taste
- blurred vision or other problems with the eye (including increased production of tears)
- dizziness
- fever; chills; shivering or headache
- signs of infection such as swelling, redness and increased temperature, fever, chills, sore throat or mouth ulcers
- bleeding or bruising more easily than normal
- changes in your voice or hoarseness
- difficulty speaking
- loss of body weight
- abdominal, pelvic, anal or back pain
- sudden headache and nausea (may include stiff/swollen neck, vision trouble)
- sudden pain in the stomach or back (may include fainting, hiccups, hearing loss, problems with balance)
These are the more common side effects of MVASI. Mostly these are mild.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- stomach cramps or pains
- severe or bloody diarrhoea
- bleeding from stomach or intestines which may look like coffee grounds or black sticky bowel motions (stools)
- nausea and vomiting; including vomiting blood or material that looks like coffee grounds
- coughing or spitting blood
- pain, redness, swelling and warmth over a vein which may suggest deep vein thrombosis (blood clots in the veins of legs)
- severe body pain including headaches
- loss of control of your bladder or bowels; passage of wind or bowel motions through the vagina
- severe bleeding
- problems with your wounds healing after surgery
- confusion
- seizures (fits)
- feeling of numbness or tingling in hands or feet
- dry mouth in combination with thirst and/or reduced or darkened urine
- abscesses (pus-filled sores)
- falling asleep or fainting
- problems with the heart with breathing difficulties
- chest pain
- increase in heart rate (pulse)
- shortness of breath
- sudden, extremely severe headache, nausea and vomiting
- a sudden pain in the back or stomach or feeling weak, numb or tingly down one side, like a stroke.
These are serious side effects. You may need urgent medical attention. Serious side effects are rare.
Tell your doctor or dentist if you experience pain in the mouth, teeth and/or jaw, swelling or sores inside the mouth, loosening of a tooth, or numbness or a feeling of heaviness in the jaw. These could be signs and symptoms of bone damage in the jaw (osteonecrosis).
Some side effects are more common in elderly patients. These include blood clots in the arteries, which can lead to a stroke or a heart attack. In addition, elderly patients have a higher risk of a reduction in the number of white cells in the blood and cells that help the blood clot, which can lead to infections and bleeding or bruising more easily than normal. Other side effects reported with a higher frequency in elderly patients were diarrhoea, nausea or sickness, headache, hair loss, inflammation of the mouth and throat, a feeling of numbness or tingling in the hands or feet and fatigue.
There have been reports of abnormal tube-like connections (fistulae) between internal organs and skin or other tissues that are not normally connected. You may have an increased risk of fistulae forming between the vagina and any part of the gastro-intestinal system if you are being treated with MVASI for cancer of the cervix.
There have been very rare reports of patients developing a hole in the septum of the nose, the structure that separates the nostrils. Symptoms may include nose bleeds, nasal congestion or infection, or whistling sounds when breathing.
MVASI is not approved for use in the eye. The following side effects may also occur if MVASI is injected directly into the eye:
- infection (some cases leading to blindness)
- eye pain, redness of the eye
- small particles or spots in your vision (floaters)
- seeing bright flashes of light with floaters, progressing to a loss of sight
- bleeding in the eye
- cataracts, leading to surgery of the eye lens
- serious side effects affecting other organs, which can be severe or life-threatening and lead to hospitalisation, e.g. stroke.
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list.
Ask your doctor or pharmacist if you don't understand anything in this list.
After receiving MVASI
Storage
MVASI will be stored in the pharmacy or on the hospital ward in a refrigerator at a temperature between 2-8°C.
Disposal
MVASI is for single use only. The vials should be used once only and any remaining contents should be discarded.
Product description
Availability
MVASI is available as 100 mg and 400 mg single-dose vials.
What MVASI looks like
MVASI is a clear to slightly opaque, colourless to pale yellow solution.
Ingredients
Active ingredient
Bevacizumab
Inactive ingredients
- trehalose dihydrate
- monobasic sodium phosphate monohydrate
- dibasic sodium phosphate
- Polysorbate 20
- water for injections
Distributor
MVASI is distributed in Australia by:
Amgen Australia Pty Ltd
Level 11,
10 Carrington St
Sydney NSW 2000
Ph: 1800 803 638
www.amgenmedinfo.com.au
Please check with your pharmacist for the latest Consumer Medicine Information.
Australian Registration Numbers
100 mg/4 mL AUST R 297455
400 mg/16 mL AUST R 297456
This leaflet was prepared in: February 2022
Published by MIMS April 2022


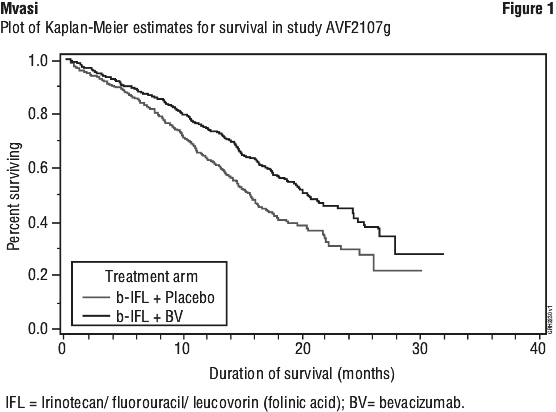
 The primary efficacy endpoint of the trial was overall survival. At the time of data cut-off, 399 deaths had occurred in patients randomised to Arm 1 (n = 225) and Arm 2 (n = 174). The addition of bevacizumab to IFL resulted in a statistically significant increase in overall survival. Results are presented in Table 4 and Figure 1. The clinical benefit of bevacizumab, as measured by survival, progression-free survival and objective response, was seen in all pre-specified patient subgroups, see Figure 2.
The primary efficacy endpoint of the trial was overall survival. At the time of data cut-off, 399 deaths had occurred in patients randomised to Arm 1 (n = 225) and Arm 2 (n = 174). The addition of bevacizumab to IFL resulted in a statistically significant increase in overall survival. Results are presented in Table 4 and Figure 1. The clinical benefit of bevacizumab, as measured by survival, progression-free survival and objective response, was seen in all pre-specified patient subgroups, see Figure 2.
 Results for the 110 patients in Arm 3 were compared to the first 100 patients enrolled in Arm 1 and Arm 2. There was a trend towards prolonged survival in the bevacizumab plus FU/LV arm as compared to the IFL plus placebo arm in this subset of patients, see Figure 3. Although the results did not show a statistical difference, the results were consistently better for the bevacizumab plus FU/LV arm than for IFL plus placebo arm for all efficacy parameters measured.
Results for the 110 patients in Arm 3 were compared to the first 100 patients enrolled in Arm 1 and Arm 2. There was a trend towards prolonged survival in the bevacizumab plus FU/LV arm as compared to the IFL plus placebo arm in this subset of patients, see Figure 3. Although the results did not show a statistical difference, the results were consistently better for the bevacizumab plus FU/LV arm than for IFL plus placebo arm for all efficacy parameters measured.

 The primary efficacy parameter of the trial was the duration of progression-free survival (PFS). In this study, there were two primary objectives: to show that XELOX was non-inferior to FOLFOX-4 and to show that bevacizumab in combination with FOLFOX-4 or XELOX chemotherapy was superior to chemotherapy alone. Both co-primary objectives were met.
The primary efficacy parameter of the trial was the duration of progression-free survival (PFS). In this study, there were two primary objectives: to show that XELOX was non-inferior to FOLFOX-4 and to show that bevacizumab in combination with FOLFOX-4 or XELOX chemotherapy was superior to chemotherapy alone. Both co-primary objectives were met. Overall response rate was similar in the chemotherapy plus bevacizumab arm (46.5%) and in chemotherapy alone arm (49.2%).
Overall response rate was similar in the chemotherapy plus bevacizumab arm (46.5%) and in chemotherapy alone arm (49.2%). No significant difference was observed in the duration of overall survival between patients who received bevacizumab monotherapy compared to patients treated with FOLFOX-4. Progression-free survival and objective response rate were inferior in the bevacizumab monotherapy arm compared to the FOLFOX-4 arm.
No significant difference was observed in the duration of overall survival between patients who received bevacizumab monotherapy compared to patients treated with FOLFOX-4. Progression-free survival and objective response rate were inferior in the bevacizumab monotherapy arm compared to the FOLFOX-4 arm.
 The efficacy and safety of bevacizumab in combination with anthracycline-based therapies have not been studied for first-line therapy in metastatic breast cancer.
The efficacy and safety of bevacizumab in combination with anthracycline-based therapies have not been studied for first-line therapy in metastatic breast cancer.
 Ninety seven patients in the IFN arm and 131 patients in the bevacizumab/IFN arm reduced the dose of IFN alfa-2a from 9 MIU to either 6 or 3 MIU, three times a week as pre-specified in the protocol.
Ninety seven patients in the IFN arm and 131 patients in the bevacizumab/IFN arm reduced the dose of IFN alfa-2a from 9 MIU to either 6 or 3 MIU, three times a week as pre-specified in the protocol. The majority of patients who were receiving steroids at baseline, including responders and non-responders, were able to reduce their steroid utilisation over time while receiving bevacizumab. The majority of patients experiencing an objective response or prolonged PFS (at week 24) were able to maintain or improve their neurocognitive function at the time of response and at week 24, respectively, compared to baseline. The majority of patients that remained in the study and were progression-free at 24 weeks, had a Karnofsky performance status (KPS) that remained stable.
The majority of patients who were receiving steroids at baseline, including responders and non-responders, were able to reduce their steroid utilisation over time while receiving bevacizumab. The majority of patients experiencing an objective response or prolonged PFS (at week 24) were able to maintain or improve their neurocognitive function at the time of response and at week 24, respectively, compared to baseline. The majority of patients that remained in the study and were progression-free at 24 weeks, had a Karnofsky performance status (KPS) that remained stable. The trial met its primary objective of PFS improvement. Compared with patients treated with chemotherapy (carboplatin and paclitaxel) alone, patients who received first-line bevacizumab at a dose of 15 mg/kg q3w in combination with chemotherapy and continued to receive bevacizumab alone had a clinically meaningful and statistically significant improvement in PFS.
The trial met its primary objective of PFS improvement. Compared with patients treated with chemotherapy (carboplatin and paclitaxel) alone, patients who received first-line bevacizumab at a dose of 15 mg/kg q3w in combination with chemotherapy and continued to receive bevacizumab alone had a clinically meaningful and statistically significant improvement in PFS.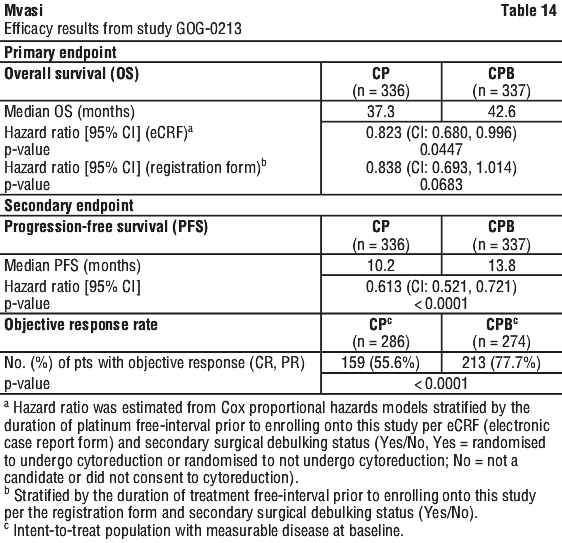 Treatment with bevacizumab at 15 mg/kg every 3 weeks in combination with chemotherapy (carboplatin + paclitaxel) for 6 and up to 8 cycles then followed by bevacizumab as a single agent resulted, when data were derived from eCRF, in a clinically meaningful and statistically significant improvement in OS compared to treatment with carboplatin + paclitaxel alone.
Treatment with bevacizumab at 15 mg/kg every 3 weeks in combination with chemotherapy (carboplatin + paclitaxel) for 6 and up to 8 cycles then followed by bevacizumab as a single agent resulted, when data were derived from eCRF, in a clinically meaningful and statistically significant improvement in OS compared to treatment with carboplatin + paclitaxel alone.

 The overall rates of discontinuation due to AEs were 8.8% in the CT arm and 43.6% in the CT + bevacizumab arm (mostly due to Grade 2-3 AEs) and the median time to discontinuation in the CT + BV arm was 5.2 months compared with 2.4 months in the CT arm. The incidence of Grade 2-5 serious AEs was 31.1% in the CT + bevacizumab arm compared with 27.1% in the CT arm. Grade 5 AEs occurred in 5 patients in the CT arm and 6 patients in the CT + bevacizumab arm with a further Grade 5 AE occurring after cross‐over to bevacizumab.
The overall rates of discontinuation due to AEs were 8.8% in the CT arm and 43.6% in the CT + bevacizumab arm (mostly due to Grade 2-3 AEs) and the median time to discontinuation in the CT + BV arm was 5.2 months compared with 2.4 months in the CT arm. The incidence of Grade 2-5 serious AEs was 31.1% in the CT + bevacizumab arm compared with 27.1% in the CT arm. Grade 5 AEs occurred in 5 patients in the CT arm and 6 patients in the CT + bevacizumab arm with a further Grade 5 AE occurring after cross‐over to bevacizumab. Interim overall efficacy results by chemotherapy backbone favoured paclitaxel and cisplatin with or without bevacizumab over paclitaxel and topotecan with or without bevacizumab, although this was not statistically significant for the primary endpoint. Median OS was 15.5 months compared to 13.3 months respectively, hazard ratio (HR) 1.15 (95% CI: 0.91, 1.46, log-rank p-Value = 0.2326), median PFS was 7.9 months compared to 5.8 months respectively, HR 1.26 (95% CI: 1.02, 1.54; log-rank p-Value = 0.0290), and the difference between ORR for the two groups was 10.9% (95% CI: 1.7, 20.1; p-Value [chi-squared] = 0.0179). An exploratory subgroup analysis for OS showed HRs for histology subgroups other than squamous-cell carcinoma that were greater than 1 (i.e. adenocarcinoma [HR = 1.17] and adenosquamous [HR = 1.03] (clinical cut-off 7 March 2014)). However the analysis was exploratory and the patient numbers in each of the histology subgroups were relatively small (adenocarcinoma n = 94 and adenosquamous carcinoma n = 44).
Interim overall efficacy results by chemotherapy backbone favoured paclitaxel and cisplatin with or without bevacizumab over paclitaxel and topotecan with or without bevacizumab, although this was not statistically significant for the primary endpoint. Median OS was 15.5 months compared to 13.3 months respectively, hazard ratio (HR) 1.15 (95% CI: 0.91, 1.46, log-rank p-Value = 0.2326), median PFS was 7.9 months compared to 5.8 months respectively, HR 1.26 (95% CI: 1.02, 1.54; log-rank p-Value = 0.0290), and the difference between ORR for the two groups was 10.9% (95% CI: 1.7, 20.1; p-Value [chi-squared] = 0.0179). An exploratory subgroup analysis for OS showed HRs for histology subgroups other than squamous-cell carcinoma that were greater than 1 (i.e. adenocarcinoma [HR = 1.17] and adenosquamous [HR = 1.03] (clinical cut-off 7 March 2014)). However the analysis was exploratory and the patient numbers in each of the histology subgroups were relatively small (adenocarcinoma n = 94 and adenosquamous carcinoma n = 44).