What is in this leaflet
This leaflet answers some common questions about Myfortic.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. Some more recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine.
You can also download the most up to date leaflet from www.novartis.com.au. Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Myfortic against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Myfortic is used for
Myfortic is used for people who have had a kidney transplant, to prevent the body from rejecting the new kidney. It is also used for people who have inflammatory kidney disease associated with a chronic autoimmune disorder known as systemic lupus erythematosis (also called lupus or SLE). Myfortic is used in combination with other medicines.
Myfortic contains the active ingredient, mycophenolic acid. It belongs to a group of medicines called immunosuppressives. These medicines work by stopping your immune system from rejecting the transplanted organ.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
Myfortic is only available with a doctor's prescription. It is not addictive.
This medicine is not recommended for use in children.
Before you take Myfortic
When you must not take it
Do not take Myfortic if you have ever had an allergic reaction to:
- mycophenolic acid, the active ingredient in Myfortic, mycophenolate sodium or mycophenolate mofetil
- any of the other ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction may include:
- rash, itching or hives on the skin
- swelling of the face, lips, tongue or other parts of the body
- faintness
- wheezing or troubled breathing
If you think you may be allergic, ask your doctor for advice without taking Myfortic.
Do not take Myfortic if you are pregnant, think you may be pregnant, or intend to become pregnant. Use of Myfortic in pregnancy causes a very high frequency of loss of pregnancy (miscarriage) and of severe birth defects in the unborn baby.
Do not take Myfortic if you are a woman who could be pregnant and you have not provided a negative pregnancy test before your first prescription.
Do not take Myfortic if you are not using effective contraception. Your doctor should advise you about contraception before you start taking Myfortic. Reliable contraception must be used before starting Myfortic. Myfortic may reduce blood levels of the hormones in oral contraceptive pills and could theoretically reduce their effectiveness. Use of two methods of contraception is advised. The two methods can be a double barrier method or a barrier method plus a hormonal method. Adequate barrier methods of contraception include: diaphragm, condom (by the partner), intrauterine device, sponge or spermicide.
Do not take Myfortic if you are breast-feeding. You must not breast-feed during treatment with Myfortic. This is because small amounts of the medicine can pass into the mother's milk.
Do not take Myfortic after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. In that case, return it to your pharmacist.
Before you start to take it
Tell your doctor if you have or have had:
- sun spots or skin cancers
- low neutrophil (a type of white blood cell) blood count
- serious problems with your stomach or bowel, such as ulcers or bleeding
- Lesch-Nyhan or Kelley-Seegmiller syndrome (a rare hereditary disorder that affects males only and is caused by a deficiency of a specific enzyme)
Your doctor may not want you to take Myfortic or may want to take special precautions if you have any of the above conditions.
Tell your doctor if you are pregnant, think you may be pregnant or plan to become pregnant You must not take Myfortic if you are pregnant. If you are a woman who could become pregnant, you must provide a negative pregnancy test before starting treatment and must follow the contraceptive advice given to you by your doctor. Your doctor may request more than one test to ensure you are not pregnant before starting Myfortic.
If you are planning to have a child, your doctor will talk to you about the risks and alternative treatments you can take to prevent rejection of your transplant organ.
Tell your doctor if you are breast-feeding.
Tell your doctor if you plan to have any vaccinations.
Tell your doctor if you are lactose intolerant. Myfortic tablets contain lactose.
Tell your doctor if you are allergic to any other medicines, foods, dyes or preservatives. Your doctor will want to know if you are prone to allergies.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines may interfere with Myfortic.
These medicines include:
- antacids that contain magnesium or aluminium hydroxide
- azathioprine, tacrolimus, or any other immunosuppressive medicine
- certain live vaccines
- cholestyramine (used to treat high blood cholesterol levels)
- aciclovir or ganciclovir (used to treat viral infections)
- oral contraceptives. These may not work as well while you are taking Myfortic and you could become pregnant. Talk to your doctor about other birth control methods that can be used while you are taking Myfortic.
You may need to take different amounts of your medicines or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking Myfortic.
If you have not told your doctor about any of these things, tell them before you take Myfortic.
How to take Myfortic
Follow all directions given to you by your doctor and pharmacist carefully. These instructions may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
Do not exceed the recommended dosage. The usual dose is 1,440 mg, taken as 720 mg twice a day. Your doctor may have prescribed a different dose, particularly for the initial treatment of inflammatory kidney disease.
Take Myfortic exactly as your doctor has prescribed. This will ensure that this medicine will work properly and prevent any unwanted side effects.
How to take it
Remove the tablets from the foil blister pack when you are ready to take them.
Swallow the tablets whole with a full glass of water.
Do not chew or crush the tablets. Do not take any tablets that are broken or split.
You may take Myfortic with or without food. It does not matter if you take Myfortic after food or on an empty stomach, as long as you take it the same way each day.
If you take it with food, always take it with food.
If you take it without food, always take it without food.
When to take it
Take Myfortic twice a day, 12 hours apart if possible.
Taking your doses 12 hours apart will have the best effect.
Take them at about the same time each day. This will help you remember when to take them.
If you have had a renal transplant, your first dose will usually be given within 48 hours after your transplant.
How long to take it
Keep taking this medicine for as long as your doctor recommends. It is important to keep taking Myfortic to prevent rejection of your transplanted kidney.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take the next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking it as you would normally.
Do not take a double dose to make up for the one that you missed. This may increase the chance of you getting an unwanted side effect.
If you miss more than one dose, contact your doctor as soon as possible.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (Overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone number 13 11 26), or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have taken too much Myfortic. Do this even if there are no signs of discomfort or poisoning.
Keep the telephone numbers for these places handy.
While you are taking Myfortic
Things you must do
Make sure that you keep all of your doctor's appointments and have any tests done that are ordered by your doctor. Your doctor may ask you to have blood tests from time to time to check your progress and prevent side effects.
Tell your doctor immediately if you have COVID-19. Increased severity of COVID-19 may occur with Myfortic.
You must not take this medicine while you are pregnant or breast-feeding. If you become pregnant while taking Myfortic, tell your doctor immediately. Your doctor will discuss the potential risks of taking Myfortic during pregnancy such as miscarriage and a higher risk of birth defects.
If you are a woman of child-bearing age, you must use effective contraception measures before you take Myfortic, while you are taking Myfortic, and for 6 weeks after you stop taking Myfortic. If you are taking oral contraceptives you must also use another form of birth control. Contact your doctor as soon as possible if you think your contraception may not have been effective or if you have forgotten to take your contraceptive pill.
If you are a sexually active man, you should use condoms during treatment with Myfortic and for 13 weeks after stopping treatment even if you have had a vasectomy. Your partner should also use effective contraception during your treatment and for 13 weeks after you have stopped Myfortic. Tell your doctor immediately if your partner becomes pregnant while you are taking Myfortic.
Do not breast-feed during treatment with Myfortic and for 6 weeks after you have stopped taking Myfortic.
If you develop lumps anywhere in your body, or develop any moles, or you notice changes in existing moles, tell your doctor. This may be an early sign of a cancer. Immunosuppressant medicines, including Myfortic, may increase the risk of developing certain cancers, including skin cancer and lymphoma (cancer of the lymphatic system).
Take special care during exposure to sunlight. If you go out in the sun, limit your exposure to sunlight and UV light by wearing a hat, protective clothing and sunscreen with a high protection factor. This will help to prevent the development of skin cancer.
Take special care of your teeth and gums. People taking immunosuppressant medicines are at a greater risk of getting infections. Taking good care of your teeth and gums will help to prevent dental and mouth infections.
If you experience any symptoms of infection (e.g. fever, sore throat), unexpected bruising and/or bleeding you should inform your doctor immediately.
If you have already had hepatitis B or C, Myfortic may increase the risk of these diseases re-appearing. Your doctor may perform blood analysis and check for symptoms of these diseases. If you experience any symptoms (yellow skin and eyes, nausea, loss of appetite, dark urine) you should inform your doctor immediately.
You must not donate blood during treatment with Myfortic and for at least 6 weeks after stopping treatment.
Men must not donate semen during treatment with Myfortic and for at least 90 days after stopping treatment.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Myfortic.
Tell any other doctor, dentist or pharmacist who treats you that you are taking Myfortic.
Things you must not do
Do not stop taking this medicine unless your doctor tells you to.
Do not have any vaccinations without first checking with your doctor. Some vaccines may be less effective or they may cause unwanted side effects while you are taking Myfortic.
Do not give this medicine to anyone else even if their condition seems similar to yours.
Do not take it to treat any other complaints unless your doctor tells you to.
Things to be careful of
Be careful driving, operating machinery or doing jobs that require you to be alert while you are taking Myfortic until you know how it affects you. This medicine is not expected to affect your ability to drive a car or operate machinery.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Myfortic, even if you do not think it is connected with the medicine. All medicines can have side effects. Sometimes they are serious, but most of the time they are not. You may need medical treatment if you get some of the side effects. Your doctor may be able to relieve some of the side effects of Myfortic by lowering the dose.
If you are over 65 years old, you should be especially careful while taking this medicine. Report any side effects promptly to your doctor. As people grow older, they are more likely to get side effects from medicines.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- diarrhoea, constipation, indigestion, flatulence, loose stools, nausea (feeling sick), or vomiting
- pain in the stomach
- pain or swelling in the joints
- weakness
- muscle pain
- indigestion, feeling bloated, or tenderness in the abdomen
- headache, dizziness
- unusual bleeding or bruising (signs of low level of blood platelets)
- cough
- anxiety
- fever (temperature above 37°C)
- tiredness
The above side effects are common.
Tell your doctor immediately if you notice any of the following:
- symptoms of infection including fever, chills, sweating, feelings of tiredness, drowsiness, or lack of energy. If you are taking Myfortic you may be more susceptible to infections than usual. These may affect various body systems, the most common being the urinary tract, the respiratory tract and the skin.
- vision changes, loss of coordination, clumsiness, memory loss, difficulty speaking or understanding what others say, and muscle weakness. These can be the signs of an infection of the brain called progressive multifocal leukoencephalopathy.
- signs of allergy such as rash, itching or hives on the skin; swelling of the face, lips, tongue or other part of the body; shortness of breath, wheezing or troubled breathing or you feel congested
- enlarged glands, a new lump or mole on your skin, or changes to existing moles, anywhere on the body. A very small number of Myfortic patients have developed cancer of the skin or lymph nodes. Lymph nodes are the bean-shaped organs found in the underarm, groin, neck, chest and abdomen that act as filters for the lymph fluid as it circulates through the body.
- any symptoms of anaemia (decrease in red blood cells), such as unusual tiredness, headache, shortness of breath at rest or with exercise, dizziness, chest pain, looking pale
- signs of peripheral oedema such as swollen hands, ankles or feet
- signs of severe high blood pressure such as headache, dizziness, possibly with nausea
- signs of high levels of potassium in the blood (hyperkalaemia) such as muscle spasms, abnormal heart rhythm
- signs of interstitial lung disease including fatal pulmonary fibrosis) such as cough, difficulty breathing, painful breathing
The above could be serious side effects that need medical attention.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed here may happen in some people. Some side effects may not give you any symptoms and can only be found when tests are done.
After using Myfortic
Storage
Keep your tablets in the foil blister pack until it is time to take them. If you take the tablets out of the blister pack, they will not keep well.
Store the pack in a cool dry place where the temperature stays below 30°C.
Always keep the tablets away from direct sunlight and keep the tablets away from moisture.
Do not store your medicine in a bathroom or near a sink.
Do not leave your medicine in the car or on window sills.
Keep the medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking Myfortic or it has passed its expiry date, ask your pharmacist what to do with any medicine you have left over.
Product description
What it looks like
Myfortic 180 mg tablets are lime green, film-coated round tablets, with bevelled edges and the imprint "C" on one side.
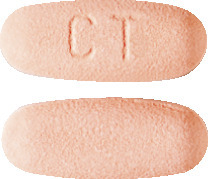
Myfortic 360 mg tablets are pale orange red film-coated ovaloid tablets with imprint 'CT' on one side.
Myfortic is available in packs of 120 tablets.
Ingredients
Active ingredient:
Myfortic gastro-resistant tablets contain either 180 mg or 360 mg mycophenolic acid per tablet, present as mycophenolate sodium
Inactive Ingredients:
Myfortic tablets also contain:
- maize starch
- povidone (E1201)
- crospovidone
- lactose
- anhydrous colloidal silica
- magnesium stearate (E572)
- hypromellose phthalate
- titanium dioxide (E171)
- iron oxide red (E172) (360 mg tablet only)
- iron oxide yellow (E172)
- indigo carmine (E132) (180 mg tablet only)
Sponsor
Myfortic is supplied in Australia by:
NOVARTIS Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
North Ryde NSW 2113
Telephone: 1 800 671 203
Web site: www.novartis.com.au
Myfortic is supplied in New Zealand by:
Novartis New Zealand Limited
PO Box 99102
Newmarket
Auckland 1149
Telephone 0800 354 335
® = Registered Trademark
This leaflet was prepared in March 2022.
Australian Registration Numbers:
Myfortic 180 mg tablet
AUST R 91939
Myfortic 360 mg tablet
AUST R 91940
Internal Document code:
(myf160322c) based on PI (myf160322i)
Published by MIMS May 2022
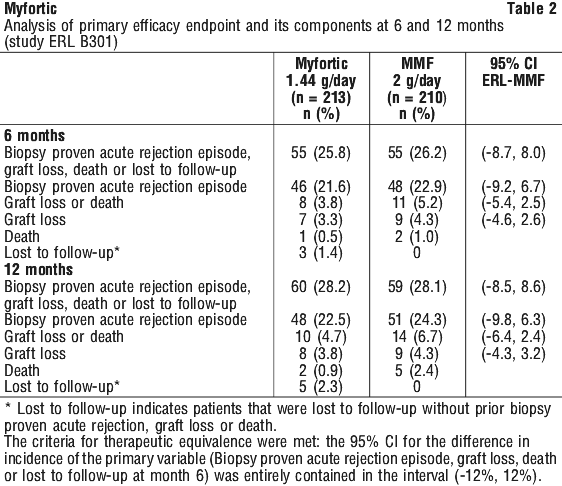
 Renal transplant patients were treated with 1440 mg Myfortic daily up to one year. A similar profile was seen in the de novo and maintenance transplant population although the incidence tended to be lower in the maintenance patients.
Renal transplant patients were treated with 1440 mg Myfortic daily up to one year. A similar profile was seen in the de novo and maintenance transplant population although the incidence tended to be lower in the maintenance patients.