Notes
Distributed by Generic Health Pty Ltd
1 Name of Medicine
Etanercept.
2 Qualitative and Quantitative Composition
Nepexto is a biosimilar medicine to Enbrel. The evidence for comparability supports the use of Nepexto for the listed indications.
Each Nepexto auto-injector contains 50 mg of etanercept and pre-filled syringe contains either 25 mg or 50 mg of etanercept.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection.
Nepexto solution for injection in the auto-injector and pre-filled syringe is a clear to opalescent, colourless to yellow solution with a pH of 6.1-6.5. The osmolality of the solution is 310 ± 30 mOsm/kg.
4.1 Therapeutic Indications
Nepexto is indicated for the treatment of:
Adults.
Rheumatoid arthritis.
Active, adult rheumatoid arthritis (RA) in patients who have had inadequate response to one or more disease-modifying antirheumatic drugs (DMARDs). Nepexto can be used in combination with methotrexate.
Severe, active rheumatoid arthritis in adults to slow progression of disease-associated structural damage in patients at high risk of erosive disease.
Psoriatic arthritis.
The signs and symptoms of active and progressive psoriatic arthritis (PsA) in adults, when the response to previous disease-modifying antirheumatic therapy has been inadequate. Etanercept has been shown to reduce the rate of progression of joint damage as measured by X-ray and to improve physical function.
Plaque psoriasis.
Adult patients with moderate to severe chronic plaque psoriasis, who are candidates for phototherapy or systemic therapy.
Ankylosing spondylitis.
The signs and symptoms of active ankylosing spondylitis (AS) in adults.
Non-radiographic axial spondyloarthritis.
Treatment of adults with active* non-radiographic axial spondyloarthritis (nr-AxSpA) with objective signs of inflammation as indicated by elevated C-reactive protein (CRP) and/or MRI change who have had an inadequate response to NSAIDs.
* Active disease is defined as a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of ≥ 4.
Children and adolescents.
Children and adolescents weighing less than 62.5 kg should not receive Nepexto. These patients should be accurately dosed on a mg/kg basis with other etanercept products.
Juvenile idiopathic arthritis.
Active polyarthritis (rheumatoid factor positive or negative) in children and adolescents, aged 2 to 17 years, who have had an inadequate response to one or more DMARDs.
Active extended oligoarthritis in children and adolescents, aged 2 to 17 years, who have had an inadequate response to, or who have proved intolerant to, methotrexate.
Active enthesitis-related arthritis in adolescents, aged 12 to 17 years, who have had an inadequate response to, or who have proved intolerant to, conventional therapy.
Active psoriatic arthritis in adolescents, aged 12 to 17 years, who have had an inadequate response to, or who have proved intolerant to, methotrexate.
Etanercept has not been studied in children aged less than 2 years.
Paediatric plaque psoriasis.
Chronic, severe plaque psoriasis in children and adolescents from 4 to 17 years, who are inadequately controlled by, or are intolerant to, other systemic therapies or phototherapies. Duration of therapy to be no longer than 24 weeks and treatment to be ceased after 12 weeks if a significant Psoriasis Area and Severity Index (PASI) response is not achieved.4.2 Dose and Method of Administration
Treatment should be initiated and supervised by specialist physicians experienced in the diagnosis and treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, plaque psoriasis or paediatric plaque psoriasis. Patients may self-inject only if their physician determines that it is appropriate and with medical follow-up, as necessary, after proper training in injection technique.
Adults.
Rheumatoid arthritis, psoriatic arthritis, non-radiographic axial spondyloarthritis and ankylosing spondylitis.
The recommended dose of Nepexto is 50 mg per week, given as a subcutaneous injection, either once weekly as a single 50 mg injection or twice weekly as two separate 25 mg injections given 3-4 days apart.
Available data in non-radiographic axial spondyloarthritis suggest a clinical response is usually achieved within 12 weeks of treatment. Continued therapy should be carefully reconsidered in a patient not responding within this time period.
Plaque psoriasis.
The recommended dose of Nepexto is 50 mg per week, given once weekly (single 50 mg injection) or twice weekly (single 25 mg injections given 3-4 days apart) as a subcutaneous injection. Higher responses may be achieved from initial treatment for up to 12 weeks with a dose of 50 mg given twice weekly, after which, the dose should be reduced to the standard dose of 50 mg per week. Treatment should be discontinued in patients who do not show a significant PASI response after 12 weeks. If re-treatment with Nepexto is indicated, the dose used should be 50 mg per week.
Elderly patients.
Elderly RA patients (age ≥ 65 years) show similar safety, efficacy and pharmacokinetic profiles compared to younger adult patients treated with Nepexto. Dose adjustment is not needed for the elderly. However, as with other medicinal products, greater sensitivity in some older patients cannot be ruled out.
Children and adolescents.
Nepexto is available as 25 mg and 50 mg pre-filled syringe, and 50 mg auto-injector.
Nepexto should only be administered in children and adolescents weighing 62.5 kg or more. The dosage of Nepexto is based on body weight for children and adolescents. Patients weighing less than 62.5 kg should be accurately dosed on a mg/kg basis using other etanercept products (see below for dosing for specific indications). Patients weighing 62.5 kg or more and receiving once weekly doses may be dosed using a 50 mg (in 1 mL) fixed-dose pre-filled syringe.
Juvenile idiopathic arthritis (age 2 years and above).
The recommended dose for children 2-17 years of age is 0.8 mg/kg (up to a maximum of 50 mg per dose) given once weekly as a subcutaneous injection, or 0.4 mg/kg (up to a maximum of 25 mg), given twice weekly with an interval of 3-4 days between doses.
Paediatric plaque psoriasis (age 4 years and above).
The recommended dose is 0.8 mg/kg (up to a maximum of 50 mg per dose), given once weekly as a subcutaneous injection for up to 24 weeks. Treatment should be discontinued in patients who do not show a significant PASI response after 12 weeks. If re-treatment with Nepexto is indicated, the above guidance on treatment duration should be followed.
Renal impairment.
No dosage adjustment is required.
Hepatic impairment.
No dosage adjustment is required.
Method of administration.
Instructions for use and handling.
Before injecting. Sites for self-injection include thigh, abdomen or upper arm. Injection sites should be rotated. New injections should be given at least 3 cm from an old site and never into areas where the skin is tender, bruised, red or hard (see instruction sheet supplied with Nepexto).
Auto-injector and pre-filled syringe (solution for injection).
Before injecting, Nepexto single-use auto-injector or pre-filled syringe should be removed from the refrigerator and allowed to reach room temperature (approximately 30 minutes). The needle cap should not be removed during this period. The solution should not be warmed in any other way. Once the solution has reached room temperature, immediate use is recommended. The solution should be clear to opalescent, colourless to yellow and the liquid may contain trace levels of translucent to white particles. The solution should not be used if it is discoloured, cloudy, or if particles other than those described are present. If the patient is concerned with the appearance of the solution, then their pharmacist should be contacted for assistance.
Nepexto is for single use only. Any unused product should be disposed of appropriately.
Administration. If a patient is to self-administer Nepexto, they should be instructed in injection techniques to ensure the safe self-administration of Nepexto (see instruction sheet supplied with Nepexto). The first injection should be performed under the supervision of a qualified health care professional. The ability of that patient to self-inject subcutaneously should be assessed. A puncture-resistant container for disposal of auto-injectors, needles and syringes should be used. Patients should be instructed in the technique and told the importance of proper auto-injector, syringe and needle disposal and be cautioned against reuse of these items.
Disposal. Contains no antimicrobial agent. Product is for single use only in one patient only. Discard any residue.
Missed dose. If a dose is missed, patients should be advised to administer the dose as soon as they remember, unless the next scheduled dose is the next day, in which case the missed dose should be skipped. Patients should continue to inject the medicine on their usual day(s). If a patient does not remember until the day that the next injection is due, instruct the patient not to take a double dose.4.3 Contraindications
Known hypersensitivity to etanercept or to any of its excipients.
Patients with, or at risk of, sepsis.
Treatment with Nepexto should not be initiated in patients with serious, active infection including chronic or localised infections.
Concurrent treatment with interleukin-1 antagonists.
4.4 Special Warnings and Precautions for Use
Traceability.
In order to improve the traceability of biological medicines, the trade name and the batch number of the administered product should be clearly recorded (or stated) in the patient file.
Infections.
Patients should be evaluated for infections before, during and after treatment with Nepexto (etanercept), taking into consideration that the mean elimination half-life of etanercept is 80 hours (standard deviation of 28 hours; range from 7 to 300 hours).
Serious infections including sepsis and tuberculosis, have been reported with the use of etanercept (see Section 4.8 Adverse Effects (Undesirable Effects)). Some of these infections have been fatal. These infections were due to bacteria, mycobacteria, fungi, viruses and parasites (including protozoa). Opportunistic infections have also been reported (including listeriosis, legionellosis and invasive fungal infections) in patients receiving etanercept. Many of these serious events have occurred in patients receiving concomitant medicines including immunosuppressants, or with underlying diseases that, in addition to their RA, could predispose them to infections. In some cases, fungal and other opportunistic infections are not recognised and this has resulted in delays in appropriate treatment, sometimes resulting in death. Patients who develop a new infection while undergoing treatment with Nepexto should be monitored closely. Administration of Nepexto should be discontinued if a patient develops a serious infection (e.g. tuberculosis or an atypical mycobacterial infection) or sepsis.
In evaluating patients for infections, physicians should consider the patient's risk for relevant opportunistic infections (e.g. exposure to endemic mycoses). Physicians should exercise caution when considering the use of Nepexto in patients with a history of recurring or chronic infections or with underlying conditions, which may predispose patients to infections such as advanced or poorly controlled diabetes (see Section 4.3 Contraindications). Caution should be exercised in patients at high risk of developing serious infection, including patients undergoing major surgery.
Tuberculosis (TB).
Tuberculosis (including disseminated or extrapulmonary presentation) has been observed in patients receiving TNF-blocking agents, including etanercept. Tuberculosis may be due to reactivation of latent TB infection or to new infection.
Before initiation of therapy with Nepexto, any patient at increased risk for TB should be evaluated for active or latent infection. If active TB is diagnosed, Nepexto therapy must not be initiated.
Prophylaxis of latent TB infection should be initiated prior to therapy with Nepexto. Treatment of latent TB in patients with a reactive tuberculin test reduces the risk of TB reactivation in patients receiving TNF blockers.
Some patients who tested negative for latent TB prior to receiving etanercept have developed active TB. Physicians should monitor patients receiving Nepexto for signs and symptoms of active TB, including patients who tested negative for latent TB infection. Applicable local guidelines should be consulted. Patients with RA appear to have an increased rate of TB infection.
Cases of TB and atypical mycobacterial infections including Mycobacterium avium complex in patients on treatment with etanercept have been reported. Treatment should be ceased immediately if mycobacterial infection is suspected.
All patients should be informed to seek medical advice if signs/symptoms suggestive of TB (e.g. persistent cough, wasting/weight loss, low grade fever) appear during or after Nepexto treatment.
Reactivation of hepatitis B.
Reactivation of hepatitis B in patients who were previously infected with the hepatitis B virus (HBV) and had received TNF blockers, including etanercept, has been reported. In some instances, HBV reactivation occurring in conjunction with TNF blocker therapy has been fatal. The majority of these reports have occurred in patients concomitantly receiving other medications that suppress the immune system, which may also contribute to HBV reactivation. Patients at risk for HBV infection should be evaluated for evidence of prior HBV infection before initiating TNF blocker therapy. Prescribers should exercise caution in prescribing TNF blockers for patients previously infected with HBV. Patients who were previously infected with HBV and require treatment with TNF blockers should be closely monitored for signs and symptoms of active HBV infection throughout therapy and for several months following termination of therapy. Adequate data are not available on the safety or efficacy of treating patients who are carriers of HBV with anti-viral therapy in conjunction with TNF blocker therapy to prevent HBV reactivation. If HBV reactivation should develop in patients who are receiving Nepexto, treatment should be stopped and effective anti-viral therapy with appropriate supportive treatment should be initiated.
Worsening of hepatitis C.
There have been reports of worsening of hepatitis C in patients receiving etanercept, although a causal relationship with etanercept has not been established.
Alcoholic hepatitis.
In a study of 48 hospitalised patients treated with etanercept or placebo for moderate to severe alcoholic hepatitis, etanercept was not efficacious and the mortality rate in patients treated with etanercept was significantly higher after 6 months. Infections were also higher in the etanercept group. The use of Nepexto in patients for the treatment of alcoholic hepatitis is not recommended. Physicians should use caution when using Nepexto in patients who also have moderate to severe alcoholic hepatitis.
Hypoglycaemia in patients treated for diabetes.
There have been reports of hypoglycaemia following initiation of etanercept in patients receiving medication for diabetes, necessitating a reduction in anti-diabetic medication in some of these patients.
Inflammatory bowel disease (IBD) and uveitis in patients with juvenile idiopathic arthritis (JIA).
There have been reports of IBD in JIA patients being treated with etanercept, which is not effective for the treatment of IBD. A causal relationship with etanercept is unclear because clinical manifestations of bowel inflammation have also been observed in untreated JIA patients. There have also been reports of uveitis in JIA patients being treated with etanercept.
Concurrent administration of TNF inhibitors and anakinra.
Concurrent administration of etanercept and anakinra (a recombinant, non-glycosylated form of the human interleukin-1 receptor antagonist) has been associated with an increased risk of serious infection, an increased risk of neutropenia and no additional benefit compared to etanercept alone. The safety and efficacy of anakinra used in combination with etanercept has not been established. Therefore, combination of Nepexto and anakinra is contraindicated (also see Section 4.3 Contraindications; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Concurrent administration of etanercept and abatacept.
In clinical studies, concurrent administration of abatacept and etanercept therapy resulted in increased incidences of serious adverse events, including infections. This combination has not demonstrated increased clinical benefit; such use is not recommended (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Interstitial lung disease.
There have been post-marketing reports of interstitial lung disease (including pneumonitis and pulmonary fibrosis), some of which had fatal outcomes.
Haematological reactions.
Rare cases of pancytopenia and very rare cases of aplastic anaemia, some with fatal outcome, have been reported in patients treated with etanercept. Caution should be exercised in patients being treated with Nepexto who have a previous history of blood dyscrasias. All patients should be advised that if they develop signs and symptoms suggestive of blood dyscrasias or infections (e.g. persistent fever, sore throat, bruising, bleeding, paleness) whilst on Nepexto, they should seek immediate medical advice. Such patients should be evaluated urgently, including full blood count; if any blood dyscrasias are confirmed, Nepexto should be discontinued.
Allergic reactions.
Parenteral administration of any biological product should be attended by appropriate precautions in case an allergic or untoward reaction occurs. Allergic reactions associated with etanercept administration have been reported commonly. Allergic reactions have included angioedema and urticaria. Serious reactions have occurred (see Section 4.8 Adverse Effects (Undesirable Effects)). If any serious allergic or anaphylactic reaction occurs, Nepexto therapy should be discontinued immediately and appropriate therapy initiated.
The rubber closure and the needle cap of the Nepexto auto-injector and pre-filled syringe do not contain any latex (dry natural rubber).
Congestive heart failure.
There have been post-marketing reports of worsening of congestive heart failure (CHF), with and without identifiable precipitating factors, in patients taking etanercept. There have also been rare (< 0.1%) reports of new onset CHF, including CHF in patients without known pre-existing cardiovascular disease. Some of these patients have been under 50 years of age. Two large clinical trials evaluating the use of etanercept in the treatment of CHF were terminated early due to lack of efficacy. Although not conclusive, data from one of these trials suggests a possible tendency towards worsening CHF and higher mortality in those patients assigned to etanercept treatment. Physicians should use caution when using Nepexto in patients who also have CHF and monitor patients carefully.
Neurological disorders.
Although no clinical trials have been performed evaluating etanercept therapy in patients with multiple sclerosis, clinical trials of other TNF antagonists in patients with multiple sclerosis have shown increases in disease activity. Treatment with etanercept and other agents that inhibit TNF have been associated with rare cases of new onset or exacerbation of central nervous system demyelinating disorders, some presenting with mental status changes and some associated with permanent disability. Cases of transverse myelitis, optic neuritis, multiple sclerosis, and new onset or exacerbation of seizure disorders have been observed in association with etanercept therapy (see Section 4.8 Adverse Effects (Undesirable Effects)). Additionally, there have been rare reports of peripheral demyelinating polyneuropathies (including Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, demyelinating polyneuropathy, and multifocal motor neuropathy). A careful risk/benefit evaluation, including a neurological assessment, is recommended when prescribing Nepexto therapy to patients with pre-existing or recent onset of central nervous system (CNS) demyelinating disease, or to those who are considered to have an increased risk of developing demyelinating disease.
Use in psoriasis.
There are limited data on the use of etanercept in combination with methotrexate for the treatment of psoriasis. The safety and efficacy of this combination in psoriasis have not been established.
The safety and efficacy of etanercept in combination with other immunosuppressive agents used in psoriasis or with phototherapy have not been studied. Nepexto should not be used in combination with such agents because of the possibility of excessive immunosuppression.
Monitoring.
Based on the results of clinical studies in rheumatoid arthritis, normally no special laboratory evaluations are necessary in addition to careful medical management and supervision of patients.
Malignancies.
Lymphomas.
TNF modulates immune responses and has a protective effect against the development of some tumours. The impact of treatment with etanercept, on the course of development of malignancies, including those caused by immunosuppressive agents, is not understood and has not been studied. The possibility exists for anti-tumour necrosis factor (TNF) therapies, including etanercept, to affect host defences against infections and malignancies since TNF mediates inflammation and modulates cellular immune responses. The impact of treatment with etanercept on the development and course of malignancies and active and/or chronic infections is not fully understood (see Section 4.8 Adverse Effects (Undesirable Effects)). Reports of malignancies affecting various sites have been received in the post-marketing period including breast and lung carcinoma and lymphoma.
In the controlled portions of clinical trials of all the TNF blocking agents, more cases of lymphoma have been observed among patients receiving the TNF blocker compared to control patients. During the controlled portions of etanercept trials, 3 lymphomas were observed among 4,509 etanercept-treated patients versus 0 among 2,040 control patients (duration of controlled treatment ranged from 3 to 24 months).
Among 6,543 adult rheumatology (RA, PsA, AS) patients treated with etanercept in controlled and uncontrolled portions of clinical trials, representing approximately 12,845 patient-years of therapy, the observed rate of lymphoma was 0.10 cases per 100 patient-years. This was 3-fold higher than the rate of lymphoma expected in the general U.S. population based on the Surveillance, Epidemiology, and End Results (SEER) Database. While patients with rheumatoid arthritis or psoriasis, particularly those with highly active disease, may be at a higher risk (up to several fold) for the development of lymphoma, a possible risk for the development of lymphomas or other malignancies in patients treated with a TNF-antagonist cannot be excluded.
Among 4,410 adult PsO patients treated with etanercept in clinical trials up to 36 months, representing approximately 4,278 patient-years of therapy, the observed rate of lymphoma was 0.05 cases per 100 patient-years, which is comparable to the rate in the general population. No cases were observed in etanercept- or placebo-treated patients during the controlled portions of these trials.
Leukaemia.
Cases of acute and chronic leukaemia have been reported in association with post-marketing TNF blocker use in rheumatoid arthritis and other indications. Even in the absence of TNF blocker therapy, patients with rheumatoid arthritis may be at higher risk (approximately 2-fold) than the general population for the development of leukaemia.
During the controlled portions of etanercept trials, 2 cases of leukaemia were observed among 5,445 (0.06 cases per 100 patient-years) etanercept-treated patients versus 0 among 2,890 control patients (duration of controlled treatment ranged from 3 to 48 months).
Among 15,401 patients treated with etanercept in controlled and open portions of clinical trials representing approximately 23,325 patient-years of therapy, the observed rate of leukaemia was 0.03 cases per 100 patient-years.
Other malignancies.
Information is available from 10,953 adult patients with 17,123 patient-years and 696 paediatric patients with 1,282 patient-years of experience across 45 etanercept clinical studies.
For malignancies other than lymphoma and non-melanoma skin cancer, there was no difference in exposure adjusted rates between the etanercept and control arms in the controlled portions of clinical studies for all indications. Analysis of the malignancy rate in combined controlled and uncontrolled portions of studies has demonstrated that types and rates are similar to what is expected in the general U.S. population based on the SEER database and suggests no increase in rates over time. Whether treatment with etanercept might influence the development and course of malignancies in adults is unknown.
Wegener's granulomatosis.
In a placebo-controlled study of 180 patients with Wegener's granulomatosis, the addition of etanercept to standard treatment (including cyclophosphamide and high-dose steroids) was no more efficacious than standard treatment alone. The group of patients who received etanercept experienced more noncutaneous malignancies of various types than the patient group receiving standard treatment alone. The use of etanercept for treatment of Wegener's granulomatosis is not recommended.
Melanoma and non-melanoma skin cancer.
Melanoma and non-melanoma skin cancer (NMSC) have been reported in patients treated with TNF-antagonists including etanercept. Post-marketing cases of Merkel cell carcinoma have been reported very infrequently in patients treated with etanercept. Periodic skin examination is recommended for all patients who are at increased risk for skin cancer. Combining the results of controlled portions of clinical trials of etanercept, more cases of non-melanoma skin cancer were observed in patients taking etanercept compared with control patients, particularly in patients with psoriasis. Long-term animal studies have not been conducted to evaluate the carcinogenic potential of etanercept.
Among 3,306 adult rheumatology (RA, PsA, AS) patients treated with etanercept in controlled clinical trials representing approximately 2,669 patient-years of therapy, the observed rate of NMSC was 0.41 cases per 100 patient-years vs 0.37 cases per 100 patient-years among 1,521 control-treated patients representing 1,077 patient-years. Among 1,245 adult psoriasis patients treated with etanercept in controlled clinical trials, representing approximately 283 patient-years of therapy, the observed rate of NMSC was 3.54 cases per 100 patient-years vs 1.28 cases per 100 patient-years among 720 control-treated patients representing 156 patient-years.
Among 15,401 patients treated with etanercept in controlled and open portions of clinical trials representing approximately 23,325 patient-years of therapy, the observed rate of melanoma was 0.043 cases per 100 patient-years.
Immunosuppression.
In a study of 49 patients with RA treated with etanercept, there was no evidence of depression of delayed-type hypersensitivity, depression of immunoglobulin levels, or change in enumeration of effector cell populations. The safety and efficacy of etanercept, in patients with immunosuppression or chronic infections have not been evaluated.
Vaccinations.
Most psoriatic patients receiving etanercept were able to mount an effective B-cell immune response to pneumococcal polysaccharide vaccine, but titres in aggregate were moderately lower and fewer patients had two-fold rises in titres compared to patients not receiving etanercept. Live vaccines should not be given concurrently with Nepexto (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). No data are available on the secondary transmission of infection by live vaccines in patients receiving etanercept. Patients with a significant exposure to varicella virus should temporarily discontinue Nepexto therapy and be considered for prophylactic treatment with varicella zoster immune globulin.
If possible, paediatric patients should be brought up to date with all immunisations (including varicella) in agreement with current immunisation guidelines prior to initiating Nepexto therapy.
Autoantibody formation.
Treatment with Nepexto may result in the formation of autoimmune antibodies (see Section 4.8 Adverse Effects (Undesirable Effects)). Rare reports have been described in clinical trials and post-marketing experience of autoimmune hepatitis, a lupus-like syndrome or rashes compatible with subacute cutaneous lupus or discoid lupus. If a patient develops symptoms and findings suggestive of autoimmune hepatitis or a lupus-like syndrome, treatment should be discontinued and the patient carefully evaluated.
Use in the elderly.
A total of 480 RA patients aged 65 years or older have been studied in clinical trials. In PsO randomised clinical trials, a total of 138 out of 1,965 patients treated with etanercept or placebo were age 65 or older. No overall differences in safety or effectiveness were observed between these patients and younger patients, but the number of geriatric PsO patients is too small to determine whether they respond differently from younger patients. The clinical trial in non-radiographic axial spondyloarthritis did not include patients aged 50 years or older. Greater sensitivity of some older individuals cannot be ruled out. Because there is a higher incidence of infections in the elderly population in general, caution should be used in treating the elderly.
Paediatric use.
Etanercept has not been studied in children less than 2 years of age. Nepexto is available as 25 mg and 50 mg pre-filled syringe, and 50 mg auto-injector.
Nepexto should only be limited to use in children and adolescents weighing 62.5 kg or more. Children and adolescents weighing less than 62.5 kg should not receive Nepexto. These patients should be accurately dosed on a mg/kg basis with other etanercept products (see Section 4.1 Therapeutic Indications; Section 4.2 Dose and Method of Administration).
Studies have not been done in patients with JIA to assess the effects of continued etanercept therapy in patients who do not respond within 3 months of initiating etanercept therapy. Additionally, studies have not been conducted to assess the effects of discontinuing or reducing the recommended dose of etanercept following its long-term use in patients with JIA.
Malignancies, some fatal, have been reported among children, adolescents and young adults who received treatment with TNF-blocking agents (initiation of therapy at ≤ 18 years of age), including etanercept to treat JIA and other indications. Approximately half of the cases were lymphomas, including Hodgkin's and non-Hodgkin's lymphoma. The other cases represented a variety of different malignancies and included rare malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months of therapy (range 1 to 84 months). Most of the patients were receiving concomitant immunosuppressants. These cases were derived from several sources including registries and post-marketing reports. In addition, there was one case of lymphoma reported in paediatric clinical trials.
Two JIA patients developed varicella infection and signs and symptoms of aseptic meningitis, which resolved without sequelae. Patients with a significant exposure to varicella virus should temporarily discontinue Nepexto therapy and be considered for prophylactic treatment with varicella zoster immune globulin.
There have been reports of inflammatory bowel disease and uveitis in patients with JIA (see Inflammatory bowel disease (IBD) and uveitis in patients with juvenile idiopathic arthritis (JIA) in this section).
The long-term effects of etanercept on the growth and development of children are not known.
Effects on laboratory tests.
No effects on laboratory tests have been reported in adults. An analysis of 54 JIA patients in an open-label study demonstrated low haemoglobin, low albumin and low lymphocyte counts in 63%, 39% and 30% of juvenile patients, respectively. These observations, however, appear to be attributed to the underlying disease, rather than treatment with etanercept.
Switching between biological therapeutics.
Limited treatment switching data is available on the safety and maintenance of treatment effect. Caution should be exercised when switching from one biological to another. The efficacy, safety/tolerability, and immunogenicity of Nepexto (Etanercept Lupin) and Enbrel were assessed in patients with moderate to severe RA despite MTX therapy (Study YLB 113-002); see Section 5.1, Clinical trials. A total of 17 subjects were involved in Stage C (a switching study whereby patients were transitioned from Nepexto to Enbrel or vice versa), with 8 subjects in the Enbrel treatment group and 9 subjects in the Nepexto treatment group.4.5 Interactions with Other Medicines and Other Forms of Interactions
Methotrexate.
Nepexto may be administered in combination with methotrexate for the treatment of rheumatoid arthritis. In a safety and efficacy trial, methotrexate had no effect on the pharmacokinetics of etanercept. The effect of etanercept on the pharmacokinetics of methotrexate has not been investigated. Product Information for methotrexate should be consulted when Nepexto is administered with methotrexate.
Abatacept.
In clinical studies, concurrent administration of abatacept and etanercept resulted in increased incidences of serious adverse events, including infections, and did not demonstrate increased clinical benefit. Use of Nepexto with abatacept is not recommended.
Anakinra.
Patients treated with etanercept and anakinra were observed to have a higher rate of serious infection (7%) when compared with patients who were treated with etanercept alone (0%, historical data). In addition, in a double-blind placebo-controlled trial, in patients receiving background methotrexate, patients treated with etanercept and anakinra were observed to have a higher rate of serious infection and neutropenia than patients who were treated with etanercept alone (see Section 4.4 Special Warnings and Precautions for Use).
Cyclophosphamide.
The use of Nepexto in patients receiving concurrent cyclophosphamide therapy is not recommended (see Section 4.4 Special Warnings and Precautions for Use, Wegener's granulomatosis).
Live vaccines.
No safety data are available on the effects of live vaccine when used in combination with etanercept. Live vaccines should therefore not be given concurrently with Nepexto.
Sulfasalazine.
In a clinical study of patients who were receiving established doses of sulfasalazine, to which etanercept was added, patients in the combination group experienced a statistically significant decrease in mean white blood cell counts in comparison to groups treated with etanercept or sulfasalazine alone.
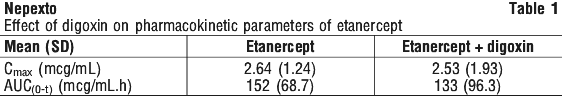
Digoxin.
Etanercept does not significantly affect digoxin exposure. There was a reduction in etanercept exposure in the presence of digoxin, however there was significant inter-subject variability. The clinical significance of this reduced exposure is uncertain. See Table 1.
Warfarin.
Etanercept does not significantly affect warfarin exposure. There was a slight reduction in etanercept exposure in the presence of warfarin, however there was significant inter-subject variability. The clinical significance of this reduced exposure is uncertain. See Table 2.

Other.
In clinical trials, no apparent interactions have been observed when etanercept was administered with glucocorticoids, salicylates (except sulfasalazine, see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions, Sulfasalazine), non-steroidal anti-inflammatory drugs (NSAIDs) or analgesics.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Long-term animal studies have not been conducted to evaluate the effects of etanercept on fertility.
(Category D)
Women of childbearing potential should be advised to use appropriate contraception to avoid becoming pregnant during Nepexto therapy and for three weeks after discontinuation of therapy.
The safe use of etanercept during pregnancy has not been established. Nepexto should only be used during pregnancy if the potential benefits to the mother outweigh the potential risks to the fetus. If Nepexto is used during pregnancy, or if the patient becomes pregnant while taking it, the woman should be advised of the possible risk to the fetus.
Developmental toxicity studies have been performed in rats and rabbits at doses resulting in AUC-based systemic exposure levels of etanercept that were at least 12-fold higher than in humans at the highest proposed therapeutic dose of 50 mg and have revealed no evidence of harm to the fetus due to etanercept. There are, however, no studies in pregnant women. Animal studies are not always predictive of human response.
Etanercept crosses the placenta and has been detected in the serum of infants born to female patients treated with etanercept during pregnancy. The clinical impact of this is unknown, however, infants may be at increased risk of infection. Administration of live vaccines to infants for 16 weeks after the mother's last dose of Nepexto is generally not recommended.
The effects of etanercept on pregnancy outcomes have been investigated in two observational cohort studies. One pregnancy registry examined the risk of major birth defects and other pregnancy outcomes in mothers with rheumatic diseases or psoriasis exposed to etanercept in the first trimester (n = 319) versus those who were unexposed to etanercept or other TNF-antagonists (n = 144). The all-inclusive odds ratio for major birth defects in those exposed to etanercept was 2.77 (95% CI 1.04-7.35) compared to non-exposed mothers with inflammatory disease. The findings showed no clear pattern of major or minor malformations. There was no increase in rates of intrauterine or postnatal growth deficits or delayed postnatal development.
In a second observational multi-country registry study comparing the risk of adverse pregnancy outcomes in women exposed to etanercept (n = 522) to those exposed to non-biologic drugs (n = 3508), there was no observed increased risk of major birth defects (adjusted odds ratio 0.96, 95% CI: 0.58-1.60). This study also showed no increased risks of minor birth defects, preterm birth, stillbirth or infections in the first year of life for infants born to women exposed to etanercept during pregnancy.
In lactating rats, following subcutaneous administration etanercept was detected in the serum of the pups. Limited information from the published literature indicates etanercept has been detected at low levels in human milk. Use of etanercept during breastfeeding should be assessed taking into account the risks and benefits of breastfeeding for the child and therapy for the mother.
While systemic exposure in a breastfed infant is expected to be low because etanercept is largely degraded in the gastrointestinal tract, limited data regarding systemic exposure in the breastfed infant are available. Therefore, the administration of live vaccines (e.g. BCG) to a breastfed infant when the mother is receiving etanercept could be considered 16 weeks after stopping breastfeeding (or at an earlier timepoint if the infant etanercept serum levels are undetectable).4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed. The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Injection site reactions.
Patients with rheumatic diseases in controlled trials treated with etanercept had a significantly higher incidence (37% cf. 10%) of injection site reactions (erythema and/or itching, pain, bleeding, bruising or swelling) compared with placebo-treated patients, and generally did not necessitate drug discontinuation. The frequency of injection site reactions was greatest in the first month and subsequently decreased in frequency. Mean duration was 3 to 5 days. No treatment was given for the majority of injection site reactions in the etanercept treatment groups, and the majority of those patients who were given treatment received topical preparations such as corticosteroids, or oral antihistamines. Some patients who experienced injection site reactions also experienced reactions at previous injection sites. In post-marketing experience, injection site bleeding and bruising have also been observed in conjunction with etanercept therapy.
In controlled trials in patients with plaque psoriasis, approximately 13.6% of patients treated with etanercept developed injection site reactions compared with 3.4% of placebo-treated patients during the first 12 weeks of treatment.
Infections.
In placebo-controlled trials, no increase in the incidence of serious infections (fatal, life-threatening, or requiring hospitalisation or intravenous antibiotics) was observed. Serious infections occurred in 6.3% of rheumatoid arthritis patients treated with etanercept for up to 48 months. These included abscess (at various sites), bacteraemia, bronchitis, bursitis, cellulitis, cholecystitis, diarrhoea, diverticulitis, endocarditis (suspected), gastroenteritis, hepatitis B, herpes zoster, leg ulcer, mouth infection, osteomyelitis, otitis, peritonitis, pneumonia, pyelonephritis, sepsis, septic arthritis, sinusitis, skin infection, skin ulcer, urinary tract infection, vasculitis, and wound infection. In the 2-year active-controlled study where patients were treated with either etanercept alone, methotrexate alone or etanercept in combination with methotrexate, the rates of serious infections were similar among the treatment groups. However, it cannot be excluded that the combination of etanercept with methotrexate could be associated with an increase in the rate of infections.
There were no differences in rates of infection among patients treated with etanercept and those treated with placebo for plaque psoriasis in placebo-controlled trials of up to 24 weeks duration. Serious infections experienced by etanercept-treated patients included cellulitis, gastroenteritis, pneumonia, cholecystitis, osteomyelitis, gastritis, appendicitis, Streptococcal fasciitis, myositis, septic shock, diverticulitis and abscess. In the double-blind and open-label psoriatic arthritis trials, 1 patient reported a serious infection (pneumonia).
Serious and fatal infections have been reported during use of etanercept; reported pathogens include bacteria, mycobacteria (including tuberculosis), viruses and fungi. Some infections have occurred within a few weeks after initiating treatment with etanercept in patients who have underlying conditions (e.g. diabetes, congestive heart failure, history of active or chronic infections) in addition to their rheumatoid arthritis. Etanercept treatment may increase mortality in patients with established sepsis.
Opportunistic infections have been reported in association with etanercept, including invasive fungal, parasitic (including protozoal), viral (including herpes zoster), bacterial (including Listeria and Legionella), and atypical mycobacterial infections (see Section 4.4 Special Warnings and Precautions for Use). In a pooled data set of clinical trials, the overall incidence of opportunistic infections was 0.09% for the 15,402 subjects who received etanercept. The exposure-adjusted rate was 0.06 events per 100 patient-years. In post-marketing experience, approximately half of all of the case reports of opportunistic infections worldwide were invasive fungal infections. The most commonly reported invasive fungal infections included Candida, Pneumocystis, Aspergillus and Histoplasma. Invasive fungal infections accounted for more than half of the fatalities amongst patients who developed opportunistic infections. The majority of the reports with a fatal outcome were in patients with Pneumocystis pneumonia, unspecified systemic fungal infections, and aspergillosis.
Malignancies and lymphoproliferative disorders.
Reports of malignancies affecting various sites have been received in the post-marketing period. The observed rates and incidences of new malignancies in clinical trials with etanercept were similar to those expected for the population studied. Patients have been observed in clinical trials with etanercept for over five years. Among 4,462 rheumatoid arthritis patients treated with etanercept in clinical trials for a mean of 27 months (approximately 10,000 patient-years of therapy), 9 lymphomas were observed for a rate of 0.09 cases per 100 patient-years. This is 3-fold higher than the rate of lymphomas expected in the general population based on the Surveillance, Epidemiology and End Results Database. An increased rate of lymphoma up to several fold has been reported in the rheumatoid arthritis patient population and may be further increased in patients with more severe disease activity (see Section 5.3 Preclinical Safety Data, Carcinogenicity).
There have been reports of malignancies in a clinical trial of patients being treated for Wegener's granulomatosis (see Section 4.4 Special Warnings and Precautions for Use, Malignancies).
Interstitial lung disease.
In controlled clinical trials of etanercept across all indications, the frequency (incidence proportion) of interstitial lung disease in patients receiving etanercept without concomitant methotrexate was rare. In the controlled clinical trials that allowed concomitant treatment with etanercept and methotrexate, the frequency (incidence proportion) of interstitial lung disease was uncommon. There have been post-marketing reports of interstitial lung disease (including pneumonitis and pulmonary fibrosis), some of which had fatal outcomes.
Elevated liver enzymes.
In the double-blind periods of controlled clinical trials of etanercept across all indications, the frequency (incidence proportion) of adverse events of elevated liver enzymes in patients receiving etanercept without concomitant methotrexate was uncommon. In the double-blind periods of controlled clinical trials that allowed concomitant treatment with etanercept and methotrexate, the frequency (incidence proportion) of adverse events of elevated liver enzymes was common.
Autoimmune hepatitis.
In controlled clinical trials of etanercept across all indications, the frequency (incidence proportion) of autoimmune hepatitis in patients receiving etanercept without concomitant methotrexate was rare. In the controlled clinical trials that allowed concomitant treatment with etanercept and methotrexate, the frequency (incidence proportion) of autoimmune hepatitis was uncommon.
Autoantibody formation.
In controlled trials, the percentage of patients who developed new positive antinuclear antibodies (ANA) (≥ 1:40), new positive anti-double-stranded DNA antibodies and new anticardiolipin antibodies were increased compared to placebo-treated patients (11% cf. 5% respectively). The percentage of patients who developed new positive anti-double-stranded DNA antibodies was also higher by radioimmunoassay (15% of patients treated with etanercept compared to 4% of placebo-treated patients) and by Crithidia luciliae assay (3% of patients treated with etanercept compared to none of placebo-treated patients). The proportion of patients treated with etanercept who developed anticardiolipin antibodies was similarly increased compared to placebo-treated patients.
Rare reports have been described in clinical trials and post-marketing experience, including patients with rheumatoid factor positive RA, who have developed additional antibodies in conjunction with autoimmune hepatitis, a lupus-like syndrome or rashes compatible with subacute cutaneous lupus or discoid lupus by clinical presentation and biopsy (see Other adverse reactions in this section). The impact of long-term treatment with etanercept on the development of autoimmune diseases is unknown. If a patient develops symptoms and findings suggestive of a lupus-like syndrome or autoimmune hepatitis following treatment with etanercept, treatment should be discontinued and the patient should be carefully evaluated.
Psoriasis.
Cases of new onset psoriasis, including pustular psoriasis and palmoplantar psoriasis, and cases of exacerbation of pre-existing psoriasis have been reported with the use of TNF blockers, including etanercept. Many of these patients were taking concomitant immunosuppressants (e.g. methotrexate, corticosteroids). Some of these patients required hospitalisation. Most patients had improvements of their psoriasis following discontinuation of their TNF blocker. Some patients have had recurrences of the psoriasis when they were re-challenged with a different TNF blocker. Discontinuation of Nepexto should be considered for severe cases and those that do not improve or that worsen despite topical treatments.
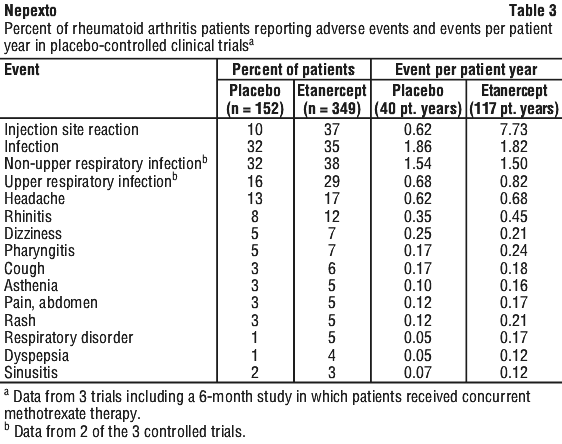
Other adverse reactions.
Events reported in at least 3% of all patients with higher incidence in patients treated with etanercept compared to controls in placebo-controlled RA trials (including the combination methotrexate trial) and events per patient-year are summarised in Table 3.
 Based on the results of clinical studies in rheumatoid arthritis, normally no special laboratory evaluations are necessary in addition to careful medical management and supervision of patients.
Based on the results of clinical studies in rheumatoid arthritis, normally no special laboratory evaluations are necessary in addition to careful medical management and supervision of patients.
The following table of suspected adverse reactions is based on clinical trials and/or spontaneous post-marketing reports.
Adverse reaction frequencies are listed in Table 4 in CIOMS frequency categories: very common: ≥ 10%; common: ≥ 1% and < 10%; uncommon: ≥ 0.1% and < 1%; rare: ≥ 0.01% and < 0.1%; very rare: < 0.01%.

Patients with non-radiographic axial spondyloarthritis.
The safety observed in adult patients with nr-AxSpA was similar to that seen in previous clinical trials of etanercept in adult patients.
Paediatric patients with juvenile idiopathic arthritis.
In general, the adverse events in paediatric patients with juvenile idiopathic arthritis were similar in frequency and type to those seen in adult patients.
JIA patients treated with etanercept had a significantly higher incidence of injection sites reactions (erythema and/or itching, pain or swelling) compared with placebo-treated patients in controlled clinical trials.
Infection was the most common adverse event reported in paediatric patients taking etanercept and occurred at an incidence similar to placebo. The types of infections reported in JIA patients were generally mild and consistent with those commonly seen in outpatient paediatric populations.
In JIA clinical trials, two cases of varicella infection with signs and symptoms suggestive of aseptic meningitis have been reported among patients treated with etanercept. There were also 4 reports of macrophage activation syndrome.
Long-term safety of etanercept monotherapy (n=103), etanercept plus methotrexate (n = 294), or methotrexate monotherapy (n = 197) were assessed for up to 3 years in a registry of 594 children aged 2 to 18 years with juvenile idiopathic arthritis, 39 of whom were 2 to 3 years of age. Overall, infections were more commonly reported in patients treated with etanercept compared to methotrexate alone (3.8% versus 2%), and the infections associated with etanercept use were of a more severe nature.
Paediatric patients with plaque psoriasis.
In a 48-week study in 211 children aged 4 to 17 years with paediatric plaque psoriasis, the adverse events reported were similar to those seen in previous studies in adults with plaque psoriasis.
Comparative safety of Nepexto and Enbrel.
The comparative safety of Nepexto and Enbrel was investigated in YLB113-002, a global phase III study in which 517 patients with moderate to severe RA received a once-weekly administration of either Nepexto or Enbrel via subcutaneous injection.
In general, Nepexto was well tolerated and showed a similar safety profile compared to the comparator drug Enbrel.
During Stage A, the randomised, double blind period of Study YLP113-002, a total of 313 subjects (60.5%) experienced at least one Treatment-Emergent Adverse Event (TEAE): 146 [55.5%] subjects in the Nepexto treatment group and 167 [65.7%] subjects in the Enbrel treatment group. The incidence of TEAEs reported as related to study medication was higher in the Enbrel treatment group (92 [36.2%] subjects) compared with the Nepexto treatment group (58 [22.1%] subjects). The number of related Serious Adverse Events (SAEs) reported was 4 (1.5%) in the Nepexto treatment group and 1 (0.4%) Enbrel group.
After completion of Stage A, subjects were given the option to continue either to Stage B (same treatment as Stage A) or Stage C (crossover of Stage A treatment) if they fulfilled the inclusion criteria for each stage. Both Stage B and Stage C were double-blind and continued in parallel.
During Stage B, a total of 269 subjects (58.0%) experienced at least one TEAE: 124 [52.8%] subjects in the Nepexto treatment group and 145 [63.3%] subjects in the Enbrel treatment group. The incidence of TEAEs reported as related to study medication was higher in the Enbrel treatment group (62 [27.1%] subjects) when compared with the Nepexto treatment group (28 [11.9%] subjects). The number of related SAEs reported was 4 (1.7%) in the Nepexto treatment group and 1 (0.4%) Enbrel group.
While the overall incidence of related SAEs are different between the groups, close examination of the events did not demonstrate any increased risk for Nepexto.
During stage C, a total of 6 subjects (33.3%) experienced at least one TEAE during the study: 3 [30.0%] subjects in the Nepexto treatment group and 3 [37.5%] subjects in the Enbrel treatment group. The incidence of TEAEs reported as related to study medication was higher in the Enbrel treatment group (2 [25.0%] subjects) when compared with the Nepexto treatment group (1 [10.0%] subjects). The number of related SAEs reported was 1 (10.0%) in the Nepexto treatment group and 2 (25.0%) Enbrel group.
No new signals were detected in the study. There were no clinically meaningful differences between treatments in terms of incidence or type of SAEs which were considered related to study medication. The overall incidence of AEs leading to discontinuation of study medication was comparable between the treatment groups, with no reports of death.
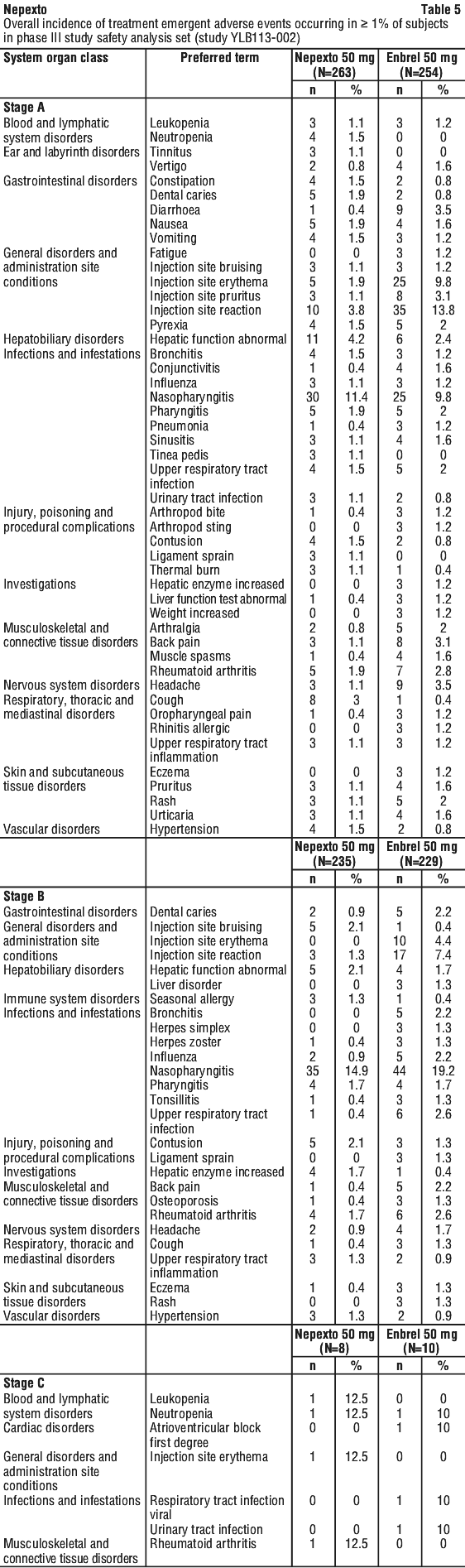
Any TEAEs that occurred in ≥ 1% of all patients who received Nepexto or Enbrel are outlined in Table 5.
Comparative safety of Nepexto and Enbrel injection site reactions and immunogenicity.
Results from the Phase 3 clinical Study YLP113-002 indicated that the incidence of reactions in the Nepexto treatment arm was significantly lower (11.0%) in comparison to those observed in the Enbrel treatment arm (31.1%). Some variation was observed between the treatment groups: injection site erythema (Nepexto 1.9% vs Enbrel 9.8%) and injection site reaction (3.8% vs 13.8%) were lower in the Nepexto treatment arm. A possible explanation for the differences may be due to the latex component in the needle shield of Enbrel, which is not a component in the needle shield of Nepexto. However, tests on the component of the needle shield have not been conducted to establish that the local site reactions are due to the presence of latex.
The maximum ADA rate reported at any time point over 52 weeks of treatment was 3.6% in the Enbrel treatment group and 0.4% in the Nepexto treatment group. The overall ADA incidence was 8.3% and 0.8% with Enbrel and Nepexto, respectively. The overall long-term immunogenicity of ADA (pooled from Stage A and B) with Nepexto was lower (0.9%) compared to Enbrel (9.2%). Most of the reported ADAs were of low titre. Of those subjects who tested positive for ADA, only 2 subjects in the Enbrel treatment arm had neutralizing ADA.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
The maximum tolerated dose of etanercept has not been established in humans. Repeat-dose studies have been performed in cynomolgus monkeys at doses resulting in AUC-based systemic exposure levels of etanercept that were over 13-fold higher than in humans at the highest proposed therapeutic dose of 50 mg and have revealed no dose-limiting or target organ toxicity. No dose-limiting toxicities were observed during clinical trials of RA patients. The highest dose level evaluated has been an IV loading dose of 32 mg/m2 followed by SC doses of 16 mg/m2 administered twice weekly. One RA patient mistakenly self-administered 62 mg etanercept SC twice weekly for three weeks without experiencing unexpected side effects. Single IV doses up to 60 mg/m2 (approximately twice the recommended dose) have been administered to healthy volunteers in an endotoxaemia study without evidence of dose-limiting toxicities.
There is no known antidote to etanercept. For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
The comparability of Nepexto with Enbrel has been demonstrated, with regard to physicochemical characteristics and efficacy and safety outcomes [see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties]. The level of comparability that has been shown supports the use of Nepexto for the listed indications.
5.1 Pharmacodynamic Properties
Etanercept binds specifically to tumour necrosis factor (TNF) and blocks its interaction with cell surface TNF receptors. Etanercept did not induce complement-mediated cytolysis of murine T-cells that expressed TNF on the cell surface. TNF is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. TNF is a dominant cytokine in the inflammatory process of rheumatoid arthritis. Elevated levels of TNF are also found in the synovium and psoriatic plaques of patients with psoriatic arthritis and in serum and synovial tissue of patients with ankylosing spondylitis. In plaque psoriasis, infiltration by inflammatory cells including T-cells leads to increased TNF levels in psoriatic lesions, compared with levels in uninvolved skin.
Two distinct receptors for TNF (TNFRs), a 55 kilodalton protein (p55) and a 75 kilodalton protein (p75), exist naturally as monomeric molecules on cell surfaces and in soluble forms. Biological activity of TNF is dependent upon binding to either cell surface TNFR.
Etanercept is a dimeric soluble form of the p75 TNF receptor that can bind to two TNF molecules. It inhibits the activity of TNF in vitro and has been shown to affect several animal models of inflammation, including murine collagen-induced arthritis. Etanercept inhibits binding of both TNFα and TNFβ (lymphotoxin alpha [LTα]) to cell surface TNFRs, rendering TNF biologically inactive. Cells expressing transmembrane TNF that bind etanercept are not lysed in vitro in the presence or absence of complement.
Mechanism of action.
Pro-inflammatory molecules that are linked in a network controlled by TNF mediate much of the joint pathology in rheumatoid arthritis and ankylosing spondylitis and skin pathology in plaque psoriasis. The mechanism of action of etanercept is thought to be its competitive inhibition of TNF binding to cell surface TNFR, preventing TNF-mediated cellular responses by rendering TNF biologically inactive. Etanercept may also modulate biological responses controlled by additional downstream molecules (e.g. cytokines, adhesion molecules, or proteinases) that are induced or regulated by TNF.
Comparability of Nepexto with Enbrel.
Comparability assessment of pharmacodynamic in vitro studies including binding and cell based assays, as well as an in vivo efficacy study in Swiss albino mice support similar/equivalent pharmacological activity of Nepexto compared to Enbrel.
The in vitro assays were closely associated with the mode of action of etanercept (including TNF-α, LT-a3 binding assays and the NF-κB reporter gene assay). In addition, Fc-related binding and functional activities were assessed, although the main function of the Fc-region of etanercept is to prolong half-life rather than to impact on Fc-mediated effector activity. Similarity between the activities of Nepexto and Enbrel were demonstrated in these in vitro studies. However, Nepexto showed higher levels of Antibody Dependent Cell Cytotoxicity (ADCC) activity than Enbrel, which is attributed to higher levels of afucosylation.
An in vivo study was conducted to demonstrate similar suppressive activity of Nepexto and Enbrel on TNF-α mediated pathology in a Swiss albino mouse model of collagen induced arthritis. Both products suppressed the development of arthritis, as determined by changes in footpad volumes and clinical scores, with no significant differences among treated groups.
Clinical trials.
This section presents data from 5 randomised controlled studies with etanercept in adults with rheumatoid arthritis, 3 studies in paediatric patients with JIA, 2 studies in adults with ankylosing spondylitis, 1 study in adults with non-radiographic axial spondyloarthritis, 1 study in adults with psoriatic arthritis and 2 studies in adults with plaque psoriasis and 1 study in paediatric patients with plaque psoriasis.
The following clinical trial information has been generated on the reference medicine.
Clinical trials with Enbrel.
Adult rheumatoid arthritis. Placebo-controlled studies.
The efficacy of etanercept was assessed in a randomised, double-blind, placebo-controlled study. The study evaluated 234 adult patients with active rheumatoid arthritis who had failed therapy with at least one but no more than four disease-modifying antirheumatic drugs (DMARDs). Doses of 10 mg or 25 mg etanercept or placebo were administered subcutaneously twice a week for 6 consecutive months. The results of this controlled trial were expressed in percentage improvement in rheumatoid arthritis using American College of Rheumatology (ACR) response criteria. The primary endpoint was achievement of an ACR 20 response at month 3. Subjects who failed to respond based on pre-specified criteria for lack of efficacy before month 3 were allowed to drop out early and were considered treatment failures. ACR 20 and 50 responses were higher in patients treated with etanercept at 3 and 6 months than in patients treated with placebo, at all time points as seen in Table 6.
 Approximately 15% of subjects who received etanercept achieved an ACR 70 response at month 3 and month 6 compared to fewer than 5% of subjects in the placebo arm. Among patients receiving etanercept, the clinical responses generally appeared within 1 to 2 weeks after initiation of therapy and nearly always occurred by 3 months. A dose response was seen; results with 10 mg were intermediate between placebo and 25 mg. Etanercept was significantly better than placebo in all components of the ACR criteria as well as other measures of rheumatoid arthritis disease activity not included in the ACR response criteria, such as morning stiffness. A Health Assessment Questionnaire (HAQ), which included disability, vitality, mental health, general health status and arthritis-associated health status sub-domains, was administered every 3 months during the trial. All sub-domains of the HAQ were improved in patients treated with etanercept compared to controls at 3 and 6 months.
Approximately 15% of subjects who received etanercept achieved an ACR 70 response at month 3 and month 6 compared to fewer than 5% of subjects in the placebo arm. Among patients receiving etanercept, the clinical responses generally appeared within 1 to 2 weeks after initiation of therapy and nearly always occurred by 3 months. A dose response was seen; results with 10 mg were intermediate between placebo and 25 mg. Etanercept was significantly better than placebo in all components of the ACR criteria as well as other measures of rheumatoid arthritis disease activity not included in the ACR response criteria, such as morning stiffness. A Health Assessment Questionnaire (HAQ), which included disability, vitality, mental health, general health status and arthritis-associated health status sub-domains, was administered every 3 months during the trial. All sub-domains of the HAQ were improved in patients treated with etanercept compared to controls at 3 and 6 months.
After discontinuation of etanercept, symptoms of arthritis generally returned within a month. Reintroduction of treatment with etanercept after discontinuations of up to 24 months resulted in the same magnitudes of response as patients who received etanercept without interruption of therapy based on results of open-label studies. Continued durable responses have been seen in open-label extension treatment trials when patients received etanercept without interruption.
A second randomised, double-blind, placebo-controlled study also compared the safety and efficacy of etanercept (25 mg) against placebo (SC, twice a week over 6 months) in 89 RA patients in addition to a stable dose of methotrexate. The ACR response criteria were used to assess efficacy. The primary endpoint was achievement of an ACR 20 response at 6 months. Responses were higher in patients treated with etanercept at 3 and 6 months. Clinical responses in etanercept-treated patients generally appeared after 1-2 weeks of therapy. In addition, approximately 15% of etanercept-treated patients achieved an ACR 70 response at month 3 and month 6, compared to less than 5% of subjects in the placebo arm. Etanercept-treated patients experienced significantly greater improvements in all components of the ACR criteria, compared to patients in the placebo arm.
The safety and efficacy of 50 mg etanercept (two 25 mg SC injections) administered once weekly were evaluated in a double-blind, placebo-controlled study of 420 patients with active RA. In this study, 53 patients received placebo, 214 patients received 50 mg etanercept once weekly and 153 patients received 25 mg etanercept twice weekly. The safety and efficacy profiles of the two etanercept treatment regimens were comparable in their effect on signs and symptoms of RA.
Active-controlled studies.
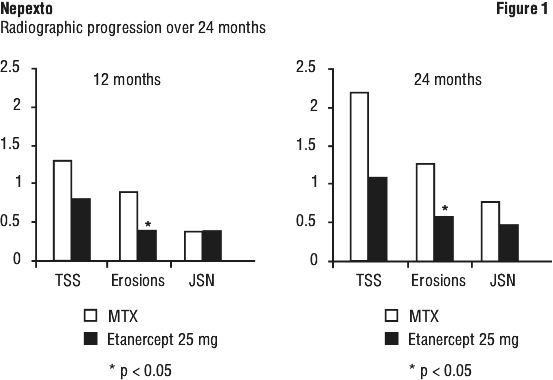
A randomised, active-controlled study with blinded radiographic evaluations as a primary endpoint compared the efficacy of etanercept to oral methotrexate in 632 adult patients with active rheumatoid arthritis (< 3 years duration) who had never received treatment with methotrexate. The patients had to have > 12 tender joints, > 10 swollen joints and either ESR > 28 mm/hr, CRP > 2.0 mg/dL, or morning stiffness for > 45 minutes. Patients were at high risk of erosive disease defined as being rheumatoid factor positive or having at least three erosions at baseline. Doses of 10 mg or 25 mg etanercept were administered SC twice a week for up to 24 months. Methotrexate doses were escalated from 7.5 mg/week to a maximum of 20 mg/week over the first 8 weeks of the trial and continued for up to 24 months. Clinical improvement including onset of action within 2 weeks with etanercept 25 mg was similar to that seen in the previous 2 trials and was maintained for up to 24 months. At baseline, patients had a moderate degree of disability, with mean HAQ scores of 1.4 to 1.5. Treatment with etanercept 25 mg resulted in substantial improvement at 12 months, with about 44% of patients achieving a normal HAQ score (less than 0.5). This benefit was maintained in Year 2 of this study.
In this study, structural joint damage was assessed radiographically and expressed as change in Total Sharp Score (TSS) and its components, the erosion score and joint space narrowing score (JSN). Radiographs of hands/wrists and feet were read at baseline and 6, 12 and 24 months. The 10 mg etanercept dose had consistently less effect on structural damage than the 25 mg dose. Etanercept 25 mg was significantly superior to methotrexate for erosion scores at both 12 and 24 months. The differences in TSS and JSN were not statistically significant between methotrexate and etanercept 25 mg. The results are shown in Figure 1.
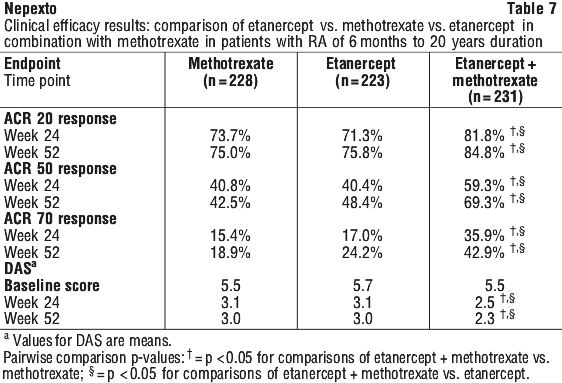
 In another active-controlled, double-blind, randomised study, clinical efficacy, safety and radiographic progression in RA patients treated with etanercept alone (25 mg twice weekly), methotrexate alone (7.5 to 20 mg weekly, median dose 20 mg) and of the combination of etanercept and methotrexate initiated concurrently were compared in 682 adult patients with active rheumatoid arthritis of 6 months to 20 years duration (median 5 years) who had a less than satisfactory response to at least 1 DMARD other than methotrexate. Forty-three percent of patients had previously received methotrexate a mean of 2 years prior to the trial at a mean dose of 12.9 mg/week. Patients were excluded from this study if methotrexate had been discontinued for lack of efficacy or for safety considerations.
In another active-controlled, double-blind, randomised study, clinical efficacy, safety and radiographic progression in RA patients treated with etanercept alone (25 mg twice weekly), methotrexate alone (7.5 to 20 mg weekly, median dose 20 mg) and of the combination of etanercept and methotrexate initiated concurrently were compared in 682 adult patients with active rheumatoid arthritis of 6 months to 20 years duration (median 5 years) who had a less than satisfactory response to at least 1 DMARD other than methotrexate. Forty-three percent of patients had previously received methotrexate a mean of 2 years prior to the trial at a mean dose of 12.9 mg/week. Patients were excluded from this study if methotrexate had been discontinued for lack of efficacy or for safety considerations.
Patients in the etanercept in combination with methotrexate therapy group had significantly higher ACR 20, ACR 50, ACR 70 responses and improvement for disease activity scores (DAS) at both 24 and 52 weeks than patients in either of the single therapy groups (results shown in Table 7).
 The percentage of patients who achieved low disease activity (defined as DAS < 2.4) at 52 weeks was 39%, 35% and 61% for patients in the etanercept alone group, methotrexate alone group and the etanercept combination group, respectively. Remission (defined as DAS < 1.6) was experienced by 18%, 14% and 37% of patients administered etanercept alone, methotrexate alone and combination therapy respectively.
The percentage of patients who achieved low disease activity (defined as DAS < 2.4) at 52 weeks was 39%, 35% and 61% for patients in the etanercept alone group, methotrexate alone group and the etanercept combination group, respectively. Remission (defined as DAS < 1.6) was experienced by 18%, 14% and 37% of patients administered etanercept alone, methotrexate alone and combination therapy respectively.
Mean HAQ scores improved from baseline levels of (1.7, 1.7 and 1.8) to (1.0, 1.1 and 0.8) at 52 weeks in the etanercept, methotrexate and etanercept in combination with methotrexate treatment groups, respectively (combination versus both methotrexate and etanercept, p < 0.01).
Radiographic progression as measured by Total Sharp Score (TSS) was significantly less in the etanercept group than in the methotrexate group at week 52. Significantly less radiographic progression (TSS) was observed with etanercept in combination with methotrexate compared with etanercept alone or methotrexate alone at week 52. The results for radiographic results (TSS), joint erosion and joint space narrowing (JSN) at week 52 are shown in Figure 2. There was a significant decrease in TSS compared with baseline in the combination of etanercept with methotrexate group.
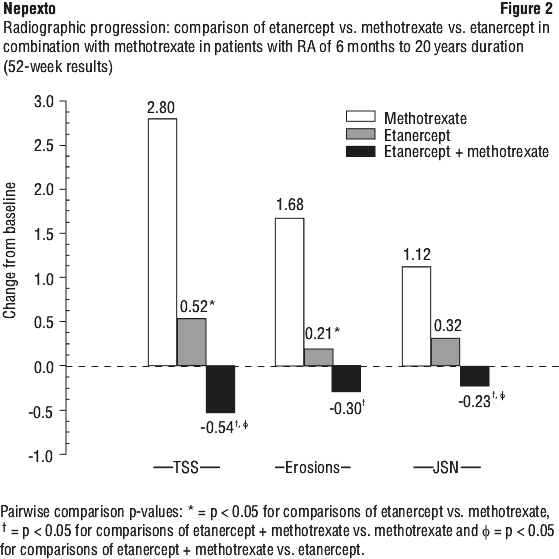 The percentage of patients without progression (TSS change ≤ 0.5) was higher in the etanercept in combination with methotrexate and etanercept groups compared with methotrexate at week 24 (74%, 68% and 56%, respectively; p < 0.05) and week 52 (80%, 68% and 57%, respectively; p < 0.05).
The percentage of patients without progression (TSS change ≤ 0.5) was higher in the etanercept in combination with methotrexate and etanercept groups compared with methotrexate at week 24 (74%, 68% and 56%, respectively; p < 0.05) and week 52 (80%, 68% and 57%, respectively; p < 0.05).
Safety, efficacy and immunogenicity were assessed in an open label study of etanercept manufactured by the serum-free process (SFP) in patients with rheumatoid arthritis. Based on indirect comparisons with historical data, the results were comparable to two previous phase 3 controlled studies in subjects with RA using etanercept manufactured by a serum-based process.
Juvenile idiopathic arthritis. The safety and efficacy of etanercept were assessed in a two-part study of 69 children with polyarticular-course juvenile idiopathic arthritis (JIA) who had a variety of JIA onset types (polyarthritis, pauciarthritis, systemic-onset). Patients aged 4 to 17 years with moderately to severely active polyarticular-course JIA refractory to or intolerant of methotrexate were enrolled; patients remained on a stable dose of a single non-steroidal anti-inflammatory drug and/or prednisone (≤ 0.2 mg/kg/day or 10 mg maximum). In part 1, all patients received 0.4 mg/kg (maximum 25 mg per dose) etanercept SC twice weekly. In part 2, patients with a clinical response at day 90 were randomised to remain on etanercept or receive placebo for four months and assessed for disease flare. Responses were measured using the ACR Pedi 30, defined as ≥ 30% improvement in at least three of six JIA core set criteria (active joint count, limitation of motion, physician and patient/parent global assessments, functional assessment and ESR) with no more than one variable worsening by more than 30%. Disease flare was defined as a ≥ 30% worsening in three of six JIA core set criteria and a minimum of two active joints. They could also have ≥ 30% improvement in not more than one of six JIA core set criteria.
In part 1 of the study, 51 of 69 (74%) patients demonstrated a clinical response and entered part 2. In part 2, 6 of 25 (24%) patients remaining on etanercept experienced a disease flare compared to 20 of 26 (77%) patients receiving placebo (p=0.007). From the start of part 2, the median time to flare was ≥ 116 days for patients who received etanercept and 28 days for patients who received placebo. Each component of the JIA core set criteria worsened in the arm that received placebo and remained stable or improved in the arm that continued on etanercept. The data suggested the possibility of a higher flare rate among those patients with a higher baseline ESR. Of patients who demonstrated a clinical response at 90 days and entered part 2 of the study, some of the patients remaining on etanercept continued to improve from month 3 through month 7, while those who received placebo did not improve.
In an open-label, safety extension study, 58 paediatric patients from the above study (from the age of 4 years at time of enrolment) continued to receive etanercept for up to 10 years. Rates of serious adverse events and serious infections did not increase with long-term exposure.
In another open-label single-arm study, 60 patients with extended oligoarthritis (15 patients aged 2 to 4, 23 patients aged 5 to 11 and 22 patients aged 12 to 17 years old), 38 patients with enthesitis-related arthritis (12 to 17 years old), and 29 patients with psoriatic arthritis (12 to 17 years old) were treated with etanercept at a dose of 0.8 mg/kg (up to a maximum of 50 mg per dose) administered weekly for 12 weeks. In each of the JIA subtypes, the majority of patients met ACR Pedi 30 criteria and demonstrated clinical improvement in secondary endpoints such as number of tender joints and physician global assessment. The safety profile was consistent with that observed in other JIA studies.
Of the 127 patients in the parent study, 109 participated in the open-label extension study and were followed for an additional 8 years for a total of up to 10 years. At the end of the extension study, 84/109 (77%) patients had completed the study; 27 (25%) while actively taking etanercept, 7 (6%) had withdrawn from treatment due to low/inactive disease; 5 (5%) had re-started etanercept following an earlier withdrawal from treatment; and 45 (41%) had stopped etanercept (but remained under observation); 25/109 (23%) patients permanently discontinued from the study. Improvements in clinical status achieved in the parent study were generally maintained for all efficacy endpoints during the entire follow-up period. Patients actively taking etanercept could enter an optional withdrawal re-treatment period once during the extension study based on investigator's judgement of clinical response. 30 patients entered the withdrawal period. Due to the small number of data points, these results should be interpreted with caution.
One malignancy, Hodgkin's disease was reported in the first year of the extension study in an 18 year old EO JIA patient. The number (exposure-adjusted rate per 100 patient-years) of serious adverse events, malignancies, and serious infections was 40 (5.85 EPl00PY), 1 (0.15 EPlO0PY), and 14 (2.05 EPl00PY), respectively. The safety profile was consistent with that observed in other JIA studies.
Long-term safety of etanercept monotherapy (n = 103), etanercept plus methotrexate (n = 294), or methotrexate monotherapy (n = 197) were assessed for up to 3 years in a registry of 594 children aged 2 to 18 years with juvenile idiopathic arthritis, 39 of whom were 2 to 3 years of age. Overall, infections were more commonly reported in patients treated with etanercept compared to methotrexate alone (3.8 versus 2%), and the infections associated with etanercept use were of a more severe nature.
Studies have not been done in patients with juvenile idiopathic arthritis to assess the effects of continued etanercept therapy in patients who do not respond within 3 months of initiating etanercept therapy. Additionally, studies have not been conducted to assess the effects of discontinuing or reducing the recommended dose of etanercept following its long-term use in patients with JIA.
The long-term effects of etanercept on the growth and development of children are not known. No formal clinical trials have been conducted in children aged 2 to 3 years. However, limited safety data from a patient registry suggest that the safety profile in children aged 2 to 3 years of age is similar to that seen in adults and children aged 4 years and older, when dosed every week with 0.8 mg/kg subcutaneously.
Adults with psoriatic arthritis. The efficacy of etanercept was assessed in a randomised, double-blind, placebo-controlled study of 205 patients with psoriatic arthritis. Patients were between 18 and 70 years of age and had active psoriatic arthritis (≥ 3 swollen joints and ≥ 3 tender joints) in at least one of the following forms: (1) distal interphalangeal (DIP) involvement; (2) polyarticular arthritis (absence of rheumatoid nodules and presence of psoriasis); (3) arthritis mutilans; (4) asymmetric psoriatic arthritis; or (5) spondylitis-like ankylosis. Patients also had plaque psoriasis with a qualifying target lesion ≥ 2 cm in diameter. Patients currently on methotrexate therapy (stable for ≥ 2 months) could continue at a stable dose of ≤ 25 mg/week methotrexate. Doses of 25 mg etanercept or placebo were administered SC twice a week for 6 months. At the end of the double-blind study, patients could enter a long-term open-label extension study for a total duration of up to 2 years.
The clinical responses were expressed as percentages of patients achieving the ACR 20, 50 and 70 response and percentages with improvement in Psoriatic Arthritis Response Criteria (PsARC). The PsARC endpoint comprises of four measures: (1) patient global assessment, (2) physician global assessment, (3) joint pain/tenderness score and (4) joint swelling score. Achievement of the PsARC endpoint requires improvement in at least two of the four measures, one of which must be joint pain/tenderness or swelling and no worsening in any of the four measures. Data have not been evaluated to establish whether etanercept inhibits progressive joint destruction in psoriatic arthritis. Results are summarised in Table 8.
 In this study, the psoriatic skin lesions of patients with active arthritis were also improved with etanercept treatment compared with placebo. In a subset of patients with psoriasis involvement ≥ 3% of body surface area, improvements in the Psoriasis Area and Severity Index (PASI) were assessed at Month 3 and Month 6. The PASI is a composite score calculated from disease activity scores and the fraction of body surface area involvement. PASI results are presented in Table 9.
In this study, the psoriatic skin lesions of patients with active arthritis were also improved with etanercept treatment compared with placebo. In a subset of patients with psoriasis involvement ≥ 3% of body surface area, improvements in the Psoriasis Area and Severity Index (PASI) were assessed at Month 3 and Month 6. The PASI is a composite score calculated from disease activity scores and the fraction of body surface area involvement. PASI results are presented in Table 9.
 Among patients with psoriatic arthritis who received etanercept, the clinical responses were apparent at the time of the first visit (4 weeks) and were maintained through 6 months of therapy. Etanercept was significantly better than placebo in all measures of disease activity (p < 0.001) and responses were similar with and without concomitant methotrexate therapy.
Among patients with psoriatic arthritis who received etanercept, the clinical responses were apparent at the time of the first visit (4 weeks) and were maintained through 6 months of therapy. Etanercept was significantly better than placebo in all measures of disease activity (p < 0.001) and responses were similar with and without concomitant methotrexate therapy.
In this study, structural joint damage was assessed radiographically and expressed as change in modified Total Sharp Score (TSS) and its components, the erosion score and joint space narrowing score (JSN). The possible range for the modified TSS was 0 to 370. Radiographs of hands and wrists were obtained at baseline and months 6, 12 and 24.
The 1-year analyses, as shown in Table 10, indicates that the difference between treatment groups was significant for mean annualised rate of change from baseline in TSS, erosion scores and for JSN. In addition, significantly more subjects in the etanercept group had no progression (≤ 0 change) in TSS from baseline, compared with subjects in the placebo group.
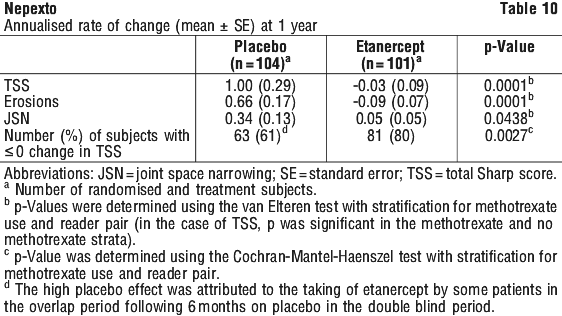 The modified TSS at 6, 12 and 24 months are presented in Table 11 for those patients who entered year 2 and provided radiographs during the second year of the study.
The modified TSS at 6, 12 and 24 months are presented in Table 11 for those patients who entered year 2 and provided radiographs during the second year of the study.
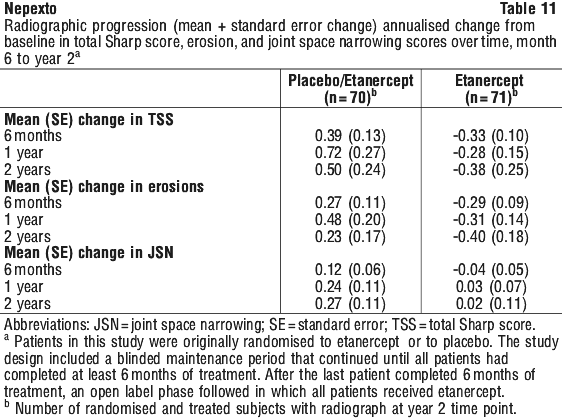 In subjects who received placebo during the controlled part of the study and etanercept in the open-label part, further radiographic progression was inhibited after subjects began receiving etanercept. Etanercept treatment resulted in improvement in physical function during the double-blind period and this benefit was maintained during the longer-term exposure of up to 2 years.
In subjects who received placebo during the controlled part of the study and etanercept in the open-label part, further radiographic progression was inhibited after subjects began receiving etanercept. Etanercept treatment resulted in improvement in physical function during the double-blind period and this benefit was maintained during the longer-term exposure of up to 2 years.
Quality of life in psoriatic arthritis patients assessed using the Health Assessment Questionnaire (HAQ) and SF-36 instruments. There was a statistically significant improvement in mean HAQ score from 1.1 to 0.5 on a scale of 0 to 3 for patients treated with etanercept. The SF-36 showed improvements in the physical but not the mental components of the quality of life score.
Adults with ankylosing spondylitis. The efficacy of etanercept was assessed in 2 randomised, double-blind, placebo-controlled studies in 361 patients with ankylosing spondylitis. The largest of these trials (n = 277) enrolled patients who were between 18 and 70 years of age and had active ankylosing spondylitis as defined by the modified New York Criteria for Ankylosing Spondylitis. Patients were to have evidence of active disease based on visual analog scale (VAS) scores of ≥ 30 for average of duration and intensity of morning stiffness plus VAS scores of ≥ 30 for at least 2 of the following 3 parameters: patient global assessment; average of VAS values for nocturnal back pain and total back pain; average of 10 questions on the Bath Ankylosing Spondylitis Functional Index (BASFI). The duration of this study was up to 24 weeks and patients had a mean diagnosis of AS for 10 years. Patients with complete ankylosis of the spine were excluded from study participation. Patients taking hydroxychloroquine, sulfasalazine, methotrexate or prednisolone (≤ 10 mg/day) or equivalent, could continue these drugs at stable doses for the duration of the study. Doses of 25 mg of etanercept (based on dose-finding studies in patients with rheumatoid arthritis) or placebo were administered subcutaneously twice a week for 6 months.
The primary measure of efficacy was a 20% improvement in the Assessment in Ankylosing Spondylitis (ASAS 20) response criteria. Compared to placebo, treatment with etanercept resulted in significant improvements in clinical response as early as 2 weeks after the initiation of therapy (see Figure 3).
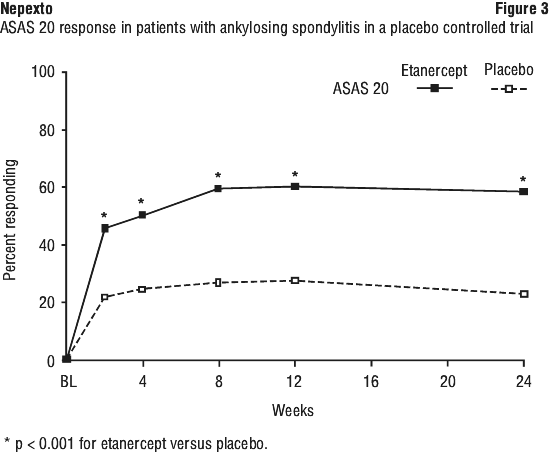 At 12 weeks, the ASAS 20/50/70 responses were achieved by 60%, 45% and 29%, respectively, of patients receiving etanercept, compared to 27%, 13% and 7%, respectively, of patients receiving placebo (p < 0.001 for etanercept vs placebo). Similar results were seen at week 24. See Table 12.
At 12 weeks, the ASAS 20/50/70 responses were achieved by 60%, 45% and 29%, respectively, of patients receiving etanercept, compared to 27%, 13% and 7%, respectively, of patients receiving placebo (p < 0.001 for etanercept vs placebo). Similar results were seen at week 24. See Table 12.
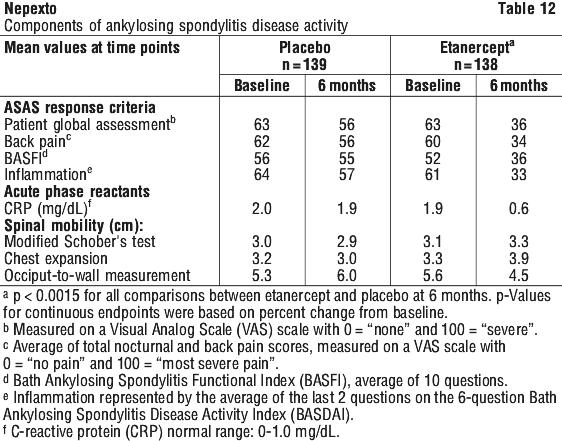 Adults with non-radiographic axial spondyloarthritis.
Adults with non-radiographic axial spondyloarthritis. Study 1.
The efficacy of etanercept in patients with non-radiographic axial spondyloarthritis (nr-AxSpA) was assessed in a randomised, 12-week double-blind, placebo-controlled study followed by an open-label period for up to an additional 92 weeks. The study evaluated 215 adult patients (modified intent-to-treat population) with active (baseline BASDAI score of ≥ 4) nr-AxSpA (18 to 49 years of age), defined as those patients meeting the ASAS classification criteria of axial spondyloarthritis but not the modified New York criteria for AS. Patients were also required to have an inadequate response or intolerance to two or more NSAIDs. In the double-blind period, patients received etanercept 50 mg weekly or placebo for 12 weeks and in the open-label period, all patients received etanercept 50 mg weekly for up to an additional 92 weeks. Throughout the study, patients were required to continue on an optimal tolerated dose of NSAIDs. The primary measure of efficacy was a 40% improvement in at least three of the four ASAS domains and absence of deterioration (ASAS 40) in the remaining domain at week 12 of the double-blind period. MRIs of the sacroiliac joint and spine and CRP were obtained to assess inflammation at baseline and at week 12. Results from the double-blind period are included below, unless noted otherwise.
Compared to placebo, treatment with etanercept resulted in statistically significant improvement in the ASAS 40, ASAS 20 and ASAS 5/6. Significant improvement was also observed for the ASAS partial remission and BASDAI 50. Week 12 results are shown in Table 13.
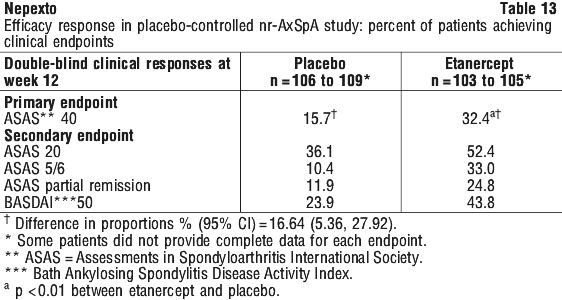 At week 12, there was an improvement in the secondary MRI endpoint SPARCC (Spondyloarthritis Research Consortium of Canada) score for the sacroiliac joint for patients receiving etanercept. Adjusted mean change from baseline was -3.8 for etanercept-treated (n = 95) versus -0.8 for placebo treated (n = 105) patients.
At week 12, there was an improvement in the secondary MRI endpoint SPARCC (Spondyloarthritis Research Consortium of Canada) score for the sacroiliac joint for patients receiving etanercept. Adjusted mean change from baseline was -3.8 for etanercept-treated (n = 95) versus -0.8 for placebo treated (n = 105) patients.
The secondary endpoints physical function and health-related quality of life were assessed using the BASFI (Bath Ankylosing Spondylitis Functional Index), EuroQol 5D and the SF-36 questionnaires. Etanercept showed greater improvement in the BASFI, EQ5D Overall Health State Score and the SF-36 Physical Component Score (PCS) from baseline to week 12 compared to placebo. Other measures of quality of life and anxiety/depression scales (including Ankylosing Spondylitis Quality of Life (ASQoL) Scores, Ankylosing Spondylitis Work Instability Index (AS-WIS) and Hospital Anxiety and Depression Scale (HADS)), and the Bath Ankylosing Spondylitis Patient Global Assessment Score (BAS-G), all secondary endpoints, showed no evidence of difference between the etanercept and placebo groups.
Clinical responses among nr-AxSpA patients who received etanercept (as assessed by ASAS 40) were apparent at the time of the first visit (2 weeks) and were maintained through 2 years of therapy. Improvements in health-related quality of life (as assessed by SF-36) and physical function (as assessed by BASFI) were also maintained through 2 years of therapy. See Figure 4.
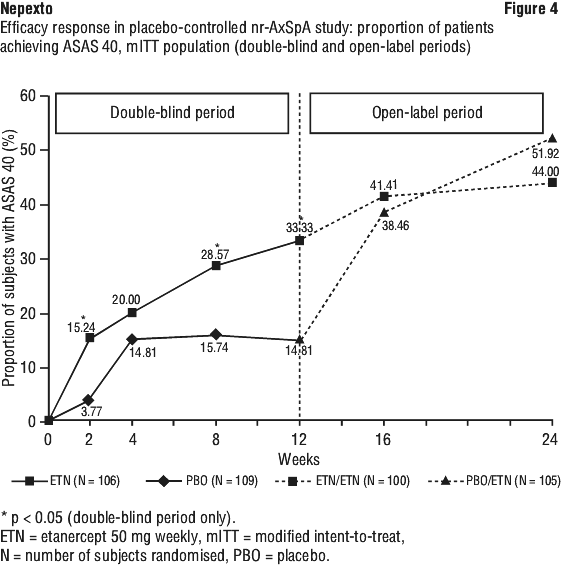 The proportions of subjects in the mITT who achieved ASAS 40 were measured at a number of time points in the open label period. At Week 12, 101 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. By Week 104, 81 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. Last observation carried forward was used to handle missing values. Based on exploratory analyses, there were no decreases in the proportions of subjects who achieved ASAS 40 at the measurement time points over the open label period compared to Week 12. There are no data on the effects of etanercept on disease progression or structural damage in nr-AxSpA patients. The 2 year data did not reveal any new safety findings.
The proportions of subjects in the mITT who achieved ASAS 40 were measured at a number of time points in the open label period. At Week 12, 101 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. By Week 104, 81 subjects who had been randomised to etanercept contributed to the ASAS 40 outcome. Last observation carried forward was used to handle missing values. Based on exploratory analyses, there were no decreases in the proportions of subjects who achieved ASAS 40 at the measurement time points over the open label period compared to Week 12. There are no data on the effects of etanercept on disease progression or structural damage in nr-AxSpA patients. The 2 year data did not reveal any new safety findings.
Study 2.
This multi-centre, open-label, phase 4, 3-period study evaluated the withdrawal and retreatment of etanercept in patients with active nr-AxSpa who achieved an adequate response (inactive disease defined as Ankylosing Spondylitis Disease Activity Score (ASDAS) C-reactive protein (CRP) less than 1.3) following 24 weeks of treatment.
209 adult patients with active nr-AxSpa (18 to 49 years of age), defined as those patients meeting the Assessment of SpondyloArthritis International Society (ASAS) classification criteria of axial spondyloarthritis (but not meeting the modified New York criteria for AS), having positive MRI findings (active inflammation on MRI highly suggestive of sacroiliitis associated with SpA) and/or positive hsCRP (defined as high sensitivity C-reactive protein [hsCRP] > 3 mg/L), and active symptoms defined by an ASDAS CRP greater than or equal to 2.1 at the screening visit received open-label etanercept 50 mg weekly plus stable background NSAID at the optimal tolerated anti-inflammatory dosage for 24 weeks in Period 1. Patients were also required to have an inadequate response or intolerance to two or more NSAIDs. At week 24, 119 (57%) patients achieved inactive disease and entered into the Period 2 40-week withdrawal phase where subjects discontinued etanercept, yet maintained the background NSAID. The primary measure of efficacy was the occurrence of flare (defined as an ASDAS erythrocyte sedimentation rate (ESR) greater than or equal to 2.1) within 40 weeks following withdrawal of etanercept. Patients who flared were retreated with etanercept 50 mg weekly for 12 weeks (Period 3).
In Period 2, the proportion of patients experiencing 2:1 flare increased from 22% (25/112) at week 4 to 67% (77/115) at week 40. Overall, 75% (86/115) patients experienced a flare at any time point within 40 weeks following withdrawal of etanercept.
The key secondary objective of Study 2 was to estimate time to flare after withdrawal of etanercept. The median time to flare following withdrawal of etanercept was 16 weeks (95% CI: 13-24 weeks). Less than 25% of patients in Study 1 who did not have treatment withdrawn experienced a flare over the equivalent 40 weeks as in Period 2 Study 2. The time to flare was shorter in subjects who discontinued etanercept treatment (Study 2) compared to subjects who received continuous etanercept treatment (Study 1).
Of the 87 patients who entered Period 3 and were retreated with etanercept 50 mg weekly for 12 weeks, 62% (54/87) reachieved inactive disease, with 50% of them reachieving it within 5 weeks (95% CI: 48 weeks).
Adults with plaque psoriasis. The safety and efficacy of etanercept were assessed in two randomised, double-blind, placebo-controlled studies. Study 1 evaluated 652 patients with chronic plaque psoriasis who were ≥ 18 years old, had active but clinically stable plaque psoriasis involving ≥ 10% of the body surface area and had a minimum psoriasis area and severity index (PASI) of 10 at screening. Etanercept was administered subcutaneously at doses of 25 mg once a week, 25 mg twice a week or 50 mg twice a week for 6 consecutive months. During the first 12 weeks of the double-blind treatment period, patients received placebo or one of the above three etanercept doses. After 12 weeks of treatment, patients in the placebo group began treatment with blinded etanercept (25 mg twice weekly); patients in the active treatment groups continued to week 24 on the dose to which they were originally randomised. This study also had a drug withdrawal period during which patients who achieved PASI improvement of at least 50% at week 24 had treatment stopped. Patients were observed off treatment for the occurrence of rebound (PASI ≥ 150% of baseline) and for the time to relapse (defined as a loss of at least half of the improvement achieved between baseline and week 24). Upon relapse, patients were retreated with etanercept in a blinded fashion at the dose they had been receiving at week 24.
Study 2 evaluated 583 patients and had the same inclusion criteria as study 1. Patients in this study received a dose of 25 mg or 50 mg etanercept, or placebo subcutaneously twice a week for 12 weeks and then all patients received open-label 25 mg etanercept twice weekly for an additional 24 weeks.
The primary efficacy endpoint in both studies was the proportion of patients in each treatment group that achieved the PASI 75 (i.e. at least a 75% improvement in the PASI score from baseline) at 12 weeks. The results of the primary and secondary endpoints of both studies are shown in Table 14.
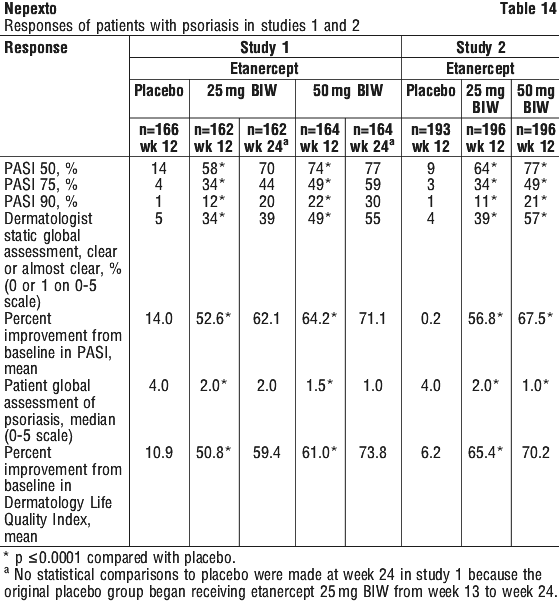 Among patients with plaque psoriasis who received etanercept, significant responses relative to placebo were apparent at the time of the first visit (2 weeks) for the mean percent improvement in PASI, Dermatologist Static Global Assessment of Psoriasis, Dermatology Life Quality Index and Patient Global Assessment of Psoriasis and were maintained through 24 weeks of therapy.
Among patients with plaque psoriasis who received etanercept, significant responses relative to placebo were apparent at the time of the first visit (2 weeks) for the mean percent improvement in PASI, Dermatologist Static Global Assessment of Psoriasis, Dermatology Life Quality Index and Patient Global Assessment of Psoriasis and were maintained through 24 weeks of therapy.
During the withdrawal period in study 1, symptoms of psoriasis gradually returned with a median time to disease relapse of 3 months. No rebound flare of disease and no psoriasis-related adverse events were observed. Retreatment with etanercept resulted in a similar magnitude of response as was seen during the initial double-blind portion of the study.
At weeks 4, 8 and 12 of study 2, the 50 mg twice weekly group had a significantly higher PASI 75 response rate than the 25 mg twice weekly group (p < 0.05, see Figure 5). The majority of patients who were initially randomised to 50 mg twice weekly and had their etanercept dose decreased at week 12 to 25 mg twice weekly maintained their PASI 75 response through week 36. For patients who received 25 mg twice weekly throughout the study, the PASI 75 response continued to improve between weeks 12 and 36.
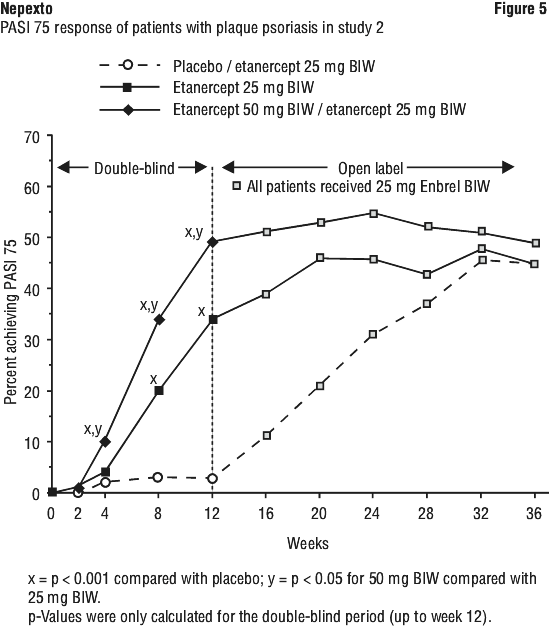 Subjects enrolled in either Study 1 or Study 2 (parent studies) were eligible to enter a phase III, open-label study to evaluate the long-term safety, tolerability, and maintenance of efficacy of etanercept in adults with plaque PsO. During the extension study, patients in one arm received etanercept 50 mg once weekly for 48 additional weeks (n = 321). See Figure 6.
Subjects enrolled in either Study 1 or Study 2 (parent studies) were eligible to enter a phase III, open-label study to evaluate the long-term safety, tolerability, and maintenance of efficacy of etanercept in adults with plaque PsO. During the extension study, patients in one arm received etanercept 50 mg once weekly for 48 additional weeks (n = 321). See Figure 6.
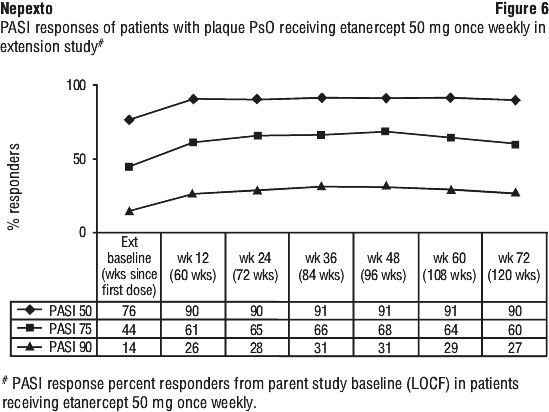 Etanercept 50 mg once-weekly continued to provide durable efficacy as demonstrated by the percentage of subjects maintaining PASI 50, 75 and 90 responses over time. It was also well tolerated in this population and its safety profile was maintained throughout the extension study.
Etanercept 50 mg once-weekly continued to provide durable efficacy as demonstrated by the percentage of subjects maintaining PASI 50, 75 and 90 responses over time. It was also well tolerated in this population and its safety profile was maintained throughout the extension study.
Paediatric patients with plaque psoriasis. The efficacy of etanercept was assessed in a randomised, double-blind, placebo-controlled study in 211 paediatric patients aged 4 to 17 years with moderate to severe plaque psoriasis (as defined by a sPGA score ≥ 3, involving ≥ 10% of the BSA, and PASI ≥ 12). Eligible patients had a history of receiving phototherapy or systemic therapy, or were inadequately controlled on topical therapy.
Patients received etanercept 0.8 mg/kg (up to 50 mg) or placebo once weekly for 12 weeks. At week 12, more patients randomised to etanercept had positive efficacy responses (e.g. PASI 75) than those randomised to placebo. See Table 15.
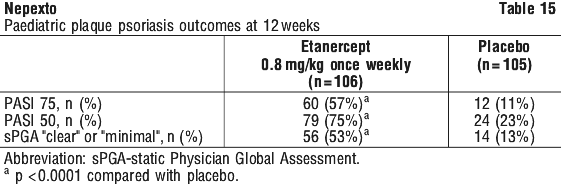 After the 12-week double-blind treatment period, all patients received etanercept 0.8 mg/kg (up to 50 mg) once weekly for an additional 24 weeks. Responses observed during the open-label period were similar to those observed in the double-blind period.
After the 12-week double-blind treatment period, all patients received etanercept 0.8 mg/kg (up to 50 mg) once weekly for an additional 24 weeks. Responses observed during the open-label period were similar to those observed in the double-blind period.
During a randomised withdrawal period, significantly more patients re-randomised to placebo experienced disease relapse (loss of PASI 75 response) compared with patients re-randomised to etanercept. With continued therapy, responses were maintained up to 48 weeks.
At week 12, the percent improvement in PASI scores from baseline was significantly higher in etanercept-treated patients compared to placebo-treated patients, across all baseline disease severity subgroups (see Table 16).
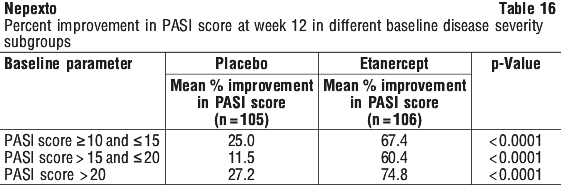 This study was conducted in children with moderate or severe psoriasis. Due to the risks associated with etanercept in children (see Section 4.4 Special Warnings and Precautions for Use), only patients with severe disease should be treated.
This study was conducted in children with moderate or severe psoriasis. Due to the risks associated with etanercept in children (see Section 4.4 Special Warnings and Precautions for Use), only patients with severe disease should be treated.
Immunocompetence. Evaluations of immunocompetence were performed on 49 etanercept-treated patients with active RA. No evidence of immunosuppression was found in evaluations of delayed-type hypersensitivity skin testing, enumeration of immune effector cell populations and immunoglobulins and in vitro testing of neutrophil and T-cell function.
Antibodies. Antibodies to etanercept, all non-neutralising, were detected in 4 out of 96 RA patients who received etanercept at a dose of 25 mg twice a week for up to 3 months in a placebo-controlled trial. Results from JIA patients were similar to those seen in adult RA patients treated with etanercept. No apparent correlation of antibody development to clinical response or adverse events was seen. Of 98 patients with psoriatic arthritis who have been tested, no patient has developed antibodies to etanercept. Among 175 ankylosing spondylitis patients treated with etanercept, 3 patients were reported with antibodies to etanercept, none were neutralising. In double-blind studies up to 6 months duration in plaque psoriasis, about 1% of the 1,084 patients developed antibodies to etanercept, none were neutralising.
Comparative efficacy of Nepexto and Enbrel. Study YLB 113-002 consisted of 517 patients between 18-75 years old with moderate to severe RA despite MTX therapy. The objective of this study was to compare the efficacy, safety / tolerability and immunogenicity of Nepexto and Enbrel at week 24 of treatment. Patients were randomised in a 1:1 ratio to receive either Nepexto 50 mg (n=266) or Enbrel 50 mg (n=262) once-weekly via subcutaneous injection along with methotrexate (MTX) in patients with active moderate to severe RA despite MTX therapy and to evaluate the long-term safety and immunogenicity of Nepexto administration. Patients enrolled in the study were followed up with up to 56 weeks after randomisation, consisting of 52 weeks of active treatment and 4 weeks of safety follow-up.
The study consisted of three stages; Stage A was considered the core study evaluating the comparative treatment efficacy of patients with moderate to severe RA. After Stage A treatment (24 weeks) and if the patients were judged eligible to enter a consecutive Stage, the patient were allocated to either Stage B or Stage C (each of 24 weeks). Stage B provided safety data including long-term immunogenicity data while Stage C was a switching study whereby patients were transitioned from Nepexto to Enbrel or vice versa.
The primary objective of Study YLB 113-002 was to demonstrate equivalence of Nepexto and Enbrel at Week 24 in terms of the American College of Rheumatology 20% response criteria (ACR20). To declare the equivalence between the treatment groups, the 2-sided 95% confidence interval of the difference in ACR20 response rates between the treatment groups should be contained within the pre-defined equivalence margin of [-15%, 15%]. Secondary objectives were to evaluate efficacy using relevant efficacy endpoints other than ACR20 at Weeks 24, and to evaluate safety/tolerability, and immunogenicity of Nepexto compared to Enbrel.
At week 24 a total number of 516 subjects with ACR20 response in each treatment group at Stage A were evaluated (Nepexto, N=263; Enbrel, N=253). Equivalence of Nepexto and Enbrel in terms of primary endpoint of ACR20 response rate at week 24 was demonstrated; response rate 81.2% for Nepexto and 86.8% for Enbrel (-5.6%), 95% CI (-11.6%, 0.5) of which was found to be within predefined equivalence margin of ±15%. Improvements in ACR20 response rates at weeks 4, 8, and 12 were also similar between treatments. The reported outcomes comprised of data from the pivotal Phase 3 study involving 517 evaluated patients for the Stage A analyses.
The time course of improvement in ACR50 and ACR70 response rates at Week 4, 8, 12, and Week 24 was comparable between treatments. An improvement in DAS28 scores from the baseline was observed over a period of time and was similar between treatment arms.
5.2 Pharmacokinetic Properties
Absorption.
Etanercept is slowly absorbed from the site of subcutaneous (SC) injection, reaching maximum concentration between 24 and 96 hours after a single dose. The absolute bioavailability is 76% as calculated in a population pharmacokinetic analysis of several studies. With twice weekly doses, it is anticipated that steady-state concentrations may be two to five-fold greater than those observed after single doses. After a single SC dose of 25 mg etanercept, the average maximum serum concentration observed in healthy volunteers was 1.65 ± 0.66 mg/L, and area under the curve was 235 ± 96.6 mg.hr/L. Dose proportionality has not been formally evaluated, but there is no apparent saturation of clearance across the dosing range.
Distribution.
A bi-exponential curve is required to describe the concentration time curve of etanercept. The central volume of distribution of etanercept is 7.6 L, while the volume of distribution at steady state is 10.4 L.
After continued dosing of RA patients (n = 25) with etanercept for 6 months with 25 mg twice weekly, the median observed level was 3.0 mg/L (range 1.7 to 5.6 mg/L).
Excretion.
Etanercept is cleared slowly from the body. The half-life is approximately 80 hours. Clearance is approximately 0.066 L/hr in patients with RA, somewhat lower than the value of 0.11 L/hr observed in healthy volunteers. Additionally, the pharmacokinetics of etanercept in rheumatoid arthritis patients, plaque psoriasis and ankylosing spondylitis patients are similar.
Serum concentration profiles at steady state were comparable among patients with RA treated with 50 mg etanercept powder for injection once weekly and those treated with 25 mg etanercept powder for injection twice weekly. A single 50 mg/mL injection of etanercept was also found to be bioequivalent to two simultaneous injections of 25 mg/mL. The mean (± standard deviation) Cmax, Cmin and partial AUC were 2.4 ± 1.5 mg/L, 1.2 ± 0.7 mg/L and 297 ± 166 mg.h/L, respectively, for patients treated with 50 mg etanercept once weekly (n = 21); and 2.6 ± 1.2 mg/L, 1.4 ± 0.7 mg/L and 316 ± 135 mg.h/L for patients treated with 25 mg etanercept twice weekly (n = 16). Serum concentrations in patients with RA have not been measured for periods of dosing that exceed 6 months. In an open-label, single-dose, two treatment crossover study in healthy volunteers, etanercept administered as a single injection of etanercept 50 mg solution for injection was found to be bioequivalent to two simultaneous injections of etanercept 25 mg powder for injection. The mean (± standard deviation) Cmax and AUC(0-t) are expressed in Table 17.
 Although there is elimination of radioactivity in urine after administration of radiolabelled etanercept to patients and volunteers, increased etanercept concentrations were not observed in patients with acute renal or hepatic failure. The presence of renal and hepatic impairment should not require a change in dosage. There is no apparent pharmacokinetic difference between men and women.
Although there is elimination of radioactivity in urine after administration of radiolabelled etanercept to patients and volunteers, increased etanercept concentrations were not observed in patients with acute renal or hepatic failure. The presence of renal and hepatic impairment should not require a change in dosage. There is no apparent pharmacokinetic difference between men and women.
No formal pharmacokinetic studies have been conducted to examine the metabolism of etanercept or the effects of renal or hepatic impairment. Methotrexate has no effect on the pharmacokinetics of etanercept. The effect of etanercept on the human pharmacokinetics of methotrexate has not been investigated.
The data described above were derived from studies using etanercept manufactured using a serum-based process.
Special populations.
Elderly (> 65 years).
The impact of advanced age was studied in the population pharmacokinetic analysis of etanercept serum concentrations. Clearance and volume estimates in patients aged 65 to 87 years were similar to estimates in patients less than 65 years of age.
Comparability of Nepexto with Enbrel.
The pharmacokinetic profiles of Nepexto and Enbrel were comparable in healthy volunteers following SC administration in study YLB 113-001 and LBC-14-155. The test/reference ratios for AUC(0-480) and Cmax were 1.12 and 1.13, in study YLB 113-001, and 1.09 and 1.05, in study LBC-14-155, for Nepexto versus Enbrel, respectively. The point estimates and 90% CIs of the ratios of AUC(0-480) and Cmax for the Nepexto/Enbrel formulations were within the interval (0.80, 1.25) demonstrating bioequivalence. Geometric mean AUC values, mean Tmax and T1/2 were comparable between Nepexto and Enbrel treatment groups as outlined in Table 18.
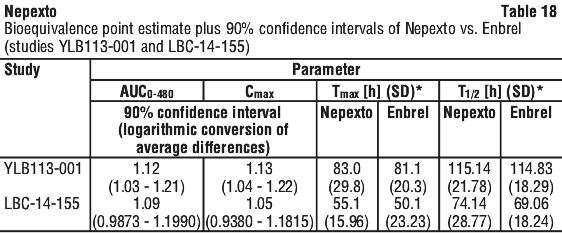
Patients with juvenile idiopathic arthritis.
In a polyarticular juvenile idiopathic arthritis (JIA) trial with etanercept, 69 patients (age 4 to 17 years) were administered 0.4 mg etanercept/kg twice weekly for three months. Serum concentration profiles were similar to those seen in adult rheumatoid arthritis patients. The youngest children (4 years of age) had reduced clearance (increased clearance when normalised by weight) compared with older children (12 years of age) and adults. Simulation of dosing suggests that while older children (10-17 years of age) will have serum levels close to those seen in adults, younger children will have appreciably lower levels.
Paediatric patients with plaque psoriasis.
Patients with paediatric plaque psoriasis (aged 4 to 17 years) were administered 0.8 mg/kg (up to a maximum dose of 50 mg per week) of etanercept once weekly for up to 48 weeks. The mean serum steady state trough concentrations ranged from 1.6 to 2.1 mg/L at weeks 12, 24, and 48. These mean concentrations in patients with paediatric plaque psoriasis were similar to the concentrations observed in patients with juvenile idiopathic arthritis (treated with 0.4 mg/kg etanercept twice weekly, up to maximum dose of 50 mg per week). These mean concentrations were similar to those seen in adult patients with plaque psoriasis treated with 25 mg etanercept twice weekly.
5.3 Preclinical Safety Data
Genotoxicity.
Genotoxicity studies showed no evidence of gene mutations or chromosomal damage.
Carcinogenicity.
Etanercept studies in animals have not been conducted to evaluate the carcinogenic potential of etanercept. See Section 4.4 Special Warnings and Precautions for Use.6 Pharmaceutical Particulars
6.1 List of Excipients
Nepexto solution for injection also contains the following excipients: sodium citrate dihydrate, monobasic sodium phosphate dihydrate, glycine, sucrose, sodium chloride, water for injections.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Solution for injection (auto-injector and pre-filled syringe).
Store at 2°C to 8°C. Refrigerate. Do not freeze.
Nepexto may be stored at temperatures up to a maximum of 25°C for a single period of up to 4 weeks, after which it should not be refrigerated again. Nepexto should be discarded if exposed to temperatures above 25°C, or if not used within 4 weeks of removal from refrigeration.
Keep Nepexto in the outer carton in order to protect from light.
6.5 Nature and Contents of Container
Auto-injector (solution for injection).
The Nepexto auto-injector is supplied in packs containing 1 or 4 single-dose pre-filled auto-injectors containing Nepexto solution. Each auto-injector contains 50 mg of the active ingredient, etanercept. The auto-injector consists of a syringe made from clear Type 1 glass with a stainless steel 27 gauge needle, needle cover cap and plastic plunger. The needle cap of the pre-filled auto-injector does not contain dry natural rubber (a derivative of latex) (see Section 4.4 Special Warnings and Precautions for Use).
Pre-filled syringe (solution for injection).
Nepexto solution for injection is supplied in packs containing 4 single-dose pre-filled glass syringes containing Nepexto solution. Each syringe of Nepexto contains either 25 mg (in 0.5 mL) or 50 mg (in 1 mL) of the active ingredient, etanercept. The syringe is made from clear Type 1 glass with a stainless steel 27 gauge needle covered with a needle cap and a stopper made of bromobutyl rubber with FluroTec coating. The needle cap does not contain natural rubber (latex) (see Section 4.4 Special Warnings and Precautions for Use).
Some strengths, pack sizes and/or pack types may not be marketed.
Australian registration numbers.
AUST R 386938 - Nepexto etanercept 50 mg in 1 mL solution for injection auto-injector.
AUST R 309834 - Nepexto etanercept 25 mg solution for injection pre-filled syringe.
AUST R 309829 - Nepexto etanercept 50 mg solution for injection pre-filled syringe.
6.6 Special Precautions for Disposal
Contains no antimicrobial agent. Product is for single use only in one patient only. Discard any residue.
In Australia, any unused medicine or waste material should be disposed of by taking it to your local pharmacy.
6.7 Physicochemical Properties
Etanercept is a human tumour necrosis factor receptor p75 Fc fusion protein produced by recombinant DNA technology in a Chinese hamster ovary (CHO) mammalian expression system. Etanercept is a dimer of a protein genetically engineered by fusing the extracellular ligand-binding domain of human tumour necrosis factor receptor-2 (TNFR2/p 75) to the Fc domain of human IgG1. This Fc component contains the hinge, CH2 and CH3 regions but not the CH1 region of IgG1. Etanercept contains 934 amino acids and has an apparent molecular weight of approximately 150 kilodaltons.
The potency is determined by measuring the ability of etanercept to neutralise the TNFα-mediated growth inhibition of A375 cells. The specific activity of etanercept is 1.7 x 106 units/mg.
CAS number.
185243-69-0.7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Summary Table of Changes
