What is in this leaflet
This leaflet answers some common questions about Nevirapine Alphapharm.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of you taking Nevirapine Alphapharm against the benefits expected for you.
If you have any concerns about taking this medicine, talk to your doctor or pharmacist.
Keep this leaflet with your medicine. You may need to read it again.
What Nevirapine Alphapharm is used for
Nevirapine Alphapharm is used in the treatment of the infection caused by the Human Immunodeficiency Virus (HIV-1).
HIV-1 is the main virus responsible for the development of Acquired Immunodeficiency Syndrome (AIDS).
Nevirapine Alphapharm contains the active ingredient Nevirapine. It belongs to a group of medicines called antiretroviral medicines called non-nucleoside reverse transcriptase inhibitors (NNRTIs).
It works by inhibiting or interrupting with the enzyme reverse transcriptase that the HIV virus needs to multiply.
Nevirapine Alphapharm does not cure or prevent HIV-1 infection or AIDS, but it does hinder the growth of HIV-1.
Nevirapine Alphapharm is used in combination with other with other antiretroviral medicines which hinder the growth of HIV-1 in other ways. When these medicines are taken with Nevirapine Alphapharm, the growth of HIV-1 is hindered more effectively.
Nevirapine Alphapharm has not been shown to reduce the likelihood that you will develop the illnesses associated with advanced HIV-1 infection.
It is important for you to continue seeing your doctor regularly.
Nevirapine Alphapharm does not reduce the risk of or prevent transmission of HIV-1 to others through sexual contact or blood contamination.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
Before you take Nevirapine Alphapharm
When you must not take it
Do not take Nevirapine Alphapharm if you have an allergy to:
- any medicine containing Nevirapine
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take this medicine if you have any rare inherited conditions of galactose and fructose intolerance.
Do not use any herbal medicines containing St John's Wort (Hypericum perforatum).
Do not take Nevirapine Alphapharm if you have or have had any of the following:
- severe liver dysfunction
- serious liver or skin reactions while on any Nevirapine treatments.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor your medical history before taking Nevirapine Alphapharm. It is essential that your doctor knows your medical history before prescribing Nevirapine Alphapharm.
Tell your doctor if you have or have had hepatitis, liver problems or liver disease.
Tell your doctor if you are having or have ever had severe kidney disease undergoing dialysis treatment.
If you are not sure if you have, or have had, any of these conditions, you should raise those concerns with your doctor.
Tell your doctor if you are pregnant or plan to become pregnant.
Your doctor can discuss with you the risks and benefits involved.
Special care is recommended during pregnancy. The benefits of Nevirapine Alphapharm must be assessed against possible effects on your unborn baby.
Tell your doctor if you are planning to breastfeed during your use of Nevirapine Alphapharm.
Breastfeeding is not recommended during your use of Nevirapine Alphapharm because:
- Nevirapine Alphapharm enters the breast milk, so your doctor may suggest an alternate method of feeding your child
- There is a risk of passing the HIV-1 virus to your baby.
If you have not told your doctor about any of the above, tell him/her before you start taking Nevirapine Alphapharm.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from a pharmacy, supermarket or health food shop.
Some medicines and Nevirapine Alphapharm may interfere with each other.
Tell your doctor if you are taking any of the following medicines:
- any other anti-HIV medicines
- anti-Hepatitis B & C medicines
- cimetidine, a medicine used to treat reflux and ulcers
- clarithromycin, an antibiotic medicine used to treat bacterial infections
- fluconazole, itraconazole or ketoconazole, medicines used to treat fungal infections
- methadone, a medicine used to treat people with opioid drug dependency
- oral contraceptives, a medicine used to prevent pregnancy
- corticosteroids (e.g. prednisone), medicines such as prednisone and cortisone, which reduce the activity of your immune system
- rifampicin or rifabutin, antibiotic medicines most commonly used to treat tuberculosis (TB)
- herbal medicines derived from St John's Wort (Hypericum perforatum)
- warfarin, a medicine used to prevent blood clots
- medicines used in the treatment of:
- allergies (antihistamines)
- bacterial/fungal infections
- cancer (e.g. cyclophosphamide monohydrate)
- depression
- epilepsy
- gastrointestinal motility disorder (e.g. cisapride)
- hypertension or heart conditions (calcium channel blockers)
- irregular heartbeats (antiarrhythmics)
- immune disorders or to prevent rejection of transplanted organ (immunosuppressants)
- migraine (ergot derivatives)
- severe pain (e.g. fentanyl)
Tell your doctor if you are taking any medicine used to treat:
- allergies, such as antihistamines
- bacterial/fungal infections
- cancer, such as cyclophosphamide
- depression
- epilepsy
- gastrointestinal motility disorder, such as cisapride
- hypertension or heart conditions, such as calcium channel blockers
- irregular heartbeats , such as antiarrhythmics
- migraine (ergot derivatives)
- severe pain, such as fentanyl.
These medicines may be affected by Nevirapine Alphapharm or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Tell your doctor if you are taking oral contraceptives. As Nevirapine Alphapharm may reduce the effectiveness of oral contraceptives, talk to your doctor about alternative methods of contraception.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take Nevirapine Alphapharm
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
Follow the dosing instructions carefully. It is important to follow the dosing instructions carefully, especially the once daily dosage during the first 14 days ('lead-in' period).
Adults 16 years and older:
- First 14 days:
one Nevirapine Alphapharm tablet once daily. - After the first 14 days:
one Nevirapine Alphapharm tablet twice daily.
Take each dose at regular 12-hour Intervals, at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
Your doctor will closely monitor you for potential side effects of taking the medicine, in particular during the first 18 weeks of treatment.
How to take it
Swallow the tablets whole with a full glass of water.
Do not crush or chew the tablets.
When to take it
It does not matter if you take this medicine before or after food.
How long to take it
Continue taking your medicine for as long as your doctor tells you to. This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose you missed.
If you have missed taking Nevirapine Alphapharm for more than 7 days, contact your doctor before you start taking it again.
You may need to restart using the 14 days (lead-in) once daily dosing procedure.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Nevirapine Alphapharm. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include:
oedema (an abnormal accumulation of fluid beneath the skin or in one or more cavities of the body) fatigue, fever, headache, insomnia, lung problems, rash, dizziness, nausea, vomiting, weight loss or erythema nodosum (a condition causing red-purple swellings on the shins, thighs and less commonly, the arms, joint and muscle pains and fever).
While you are taking Nevirapine Alphapharm
Things you must do
Contact your doctor if you experience a rash on any part of the body.
Contact your doctor immediately if the rash is accompanied by other symptoms such as fever, blisters, mouth sores, conjunctivitis, facial swelling, muscle or joint aches, swollen lymph glands, or tiredness. These may be symptoms of a hypersensitivity reaction that requires urgent medical attention.
Contact your doctor if you experience any symptoms of liver problems, such as loss of appetite, nausea, vomiting, jaundice (yellowing of the skin and/or eyes), dark coloured urine, pale coloured stools, pain/ache or sensitivity to touch in your right abdominal area (below your ribs). These could be signs of serious liver dysfunction which your doctor will need to monitor closely and may require stopping treatment with Nevirapine Alphapharm.
Keep all of your doctor's appointments so that your progress can be checked.
Liver function tests should be performed at regular intervals, especially during the first 18 weeks of treatment with Nevirapine Alphapharm. If the results are abnormal, your doctor will consider either performing more frequent liver function tests (in less severe cases) or stopping treatment with Nevirapine Alphapharm altogether (in more severe cases).
Women and patients with higher CD4 cell counts seem to be at increased risk for developing liver problems while taking Nevirapine Alphapharm.
In rare instances, temporary weakness or pain of muscles has been seen in Nevirapine Alphapharm patients experiencing skin and/or liver problems.
If you have had a previous opportunistic infection, and you notice symptoms of inflammation occurring when you first start taking Nevirapine Alphapharm, tell your doctor immediately. Symptoms of inflammation include redness, swelling, heat and pain. These symptoms have been reported in some patients who have previously had an infection when combination antiretroviral therapy was started.
If you are taking oral contraceptives (to prevent pregnancy), you should use additional or different type of contraception. Nevirapine Alphapharm may reduce effectiveness of oral contraceptives.
If you become pregnant while taking Nevirapine Alphapharm, tell your doctor immediately.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Nevirapine Alphapharm.
Things you must not do
Do not take Nevirapine Alphapharm to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or change the dosage without first checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how Nevirapine Alphapharm affects you. This medicine may cause sleepiness or drowsiness in some people. If you have these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Nevirapine Alphapharm.
This medicine helps most people with HIV-1, but it may have unwanted side effects in some people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
It may be difficult to tell whether side effects are the result of taking Nevirapine Alphapharm, effects of the HIV disease or side effects of other medicines you may be taking.
For this reason, it is very important to inform your doctor of any change in your condition.
Your doctor may need to change your dose or advise you to stop taking Nevirapine Alphapharm.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- a mild to moderate rash on the trunk, face, arms and/or legs
- fever
- nausea
- headache
- fatigue
- sleepiness or drowsiness
- abnormal liver function tests
- vomiting
- diarrhoea
- stomach pain
- myalgia (aching muscles, muscle tenderness or weakness, not caused by exercise).
The above list includes the more common side effects of your medicine.
Tell your doctor as soon as possible if you notice any of the following:
- yellowing of the skin and/or eyes, also called jaundice, hepatitis, severe and life threatening liver dysfunction (including fulminant hepatitis and liver function failure).
Combination antiretroviral therapy may cause changes in body shape, due to changes in fat distribution, in some patients. These may include:
- loss of fat from legs, arms and face
- increased fat in the abdomen and other internal organs
- breast enlargement
- fatty lumps on the back of the neck.
The above list includes serious side effects that may require medical attention.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- signs of an allergic reaction, such as shortness of breath, wheezing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin
- a severe skin reaction which starts with painful red areas, then large blisters and ends with peeling layers of skin. This is accompanied by fever and chills, aching muscles and generally feeling unwell
- severe blisters and bleeding in the lips, eyes, mouth, nose and genitals
- rash accompanied by other side effects such as fever, blisters, mouth sores, conjunctivitis, facial swelling, muscle or joint aches, swollen lymph glands, or tiredness
- signs of liver dysfunction or hepatitis (liver disease), such as nausea, vomiting, loss of appetite, feeling generally unwell, fever, itching, yellowing of the skin and eyes, light coloured bowel motions or dark coloured urine.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After taking Nevirapine Alphapharm
Storage
Keep your tablets in the pack/bottle until it is time to take them. If you take the tablets out of the pack/bottle they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 25°C.
Do not store Nevirapine Alphapharm or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Nevirapine Alphapharm 200 mg tablets are white to off white, oval shaped, biconvex uncoated tablets, debossed with "NE 200" with a single bisect separating the "NE" and "200" on one side and debossed with "M" with a single bisect separating the "M" on the other side.
Nevirapine Alphapharm is available in bottles of 60 tablets.
Ingredients
Nevirapine Alphapharm contains 200 mg of nevirapine as the active ingredient.
It also contains the following inactive ingredients:
- lactose
- microcrystalline cellulose
- povidone
- sodium starch glycollate
- colloidal anhydrous silica
- magnesium stearate
Manufacturer/ Distributor/ Supplier
Nevirapine Alphapharm is supplied in Australia by:
Alphapharm Pty Ltd
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.mylan.com.au
Australian registration numbers:
Nevirapine Alphapharm 200 mg tablets:
Bottle - AUST R 167307
This leaflet was prepared on 29 October 2019
Nevirapine AF_cmi\Oct 19/00
Published by MIMS November 2022



 Combinations of mutations were observed in nine of the 12 patients. These data from INCAS illustrate that the use of highly active drug therapies is associated with a delay in the development of antiretroviral drug resistance.
Combinations of mutations were observed in nine of the 12 patients. These data from INCAS illustrate that the use of highly active drug therapies is associated with a delay in the development of antiretroviral drug resistance.




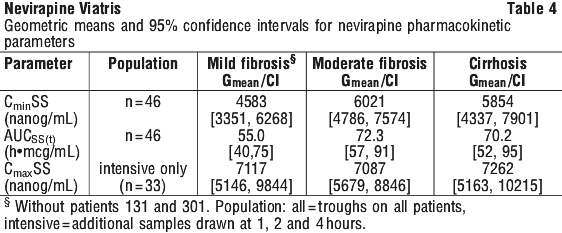 In a 200 mg nevirapine single dose pharmacokinetic study of HIV negative patients with mild and moderate hepatic impairment (Child-Pugh A, n = 6; Child-Pugh B, n = 4), a significant increase in the AUC of nevirapine was observed in one Child-Pugh B patient with ascites suggesting that patients with worsening hepatic function and ascites may be at risk of accumulating nevirapine in the systemic circulation. Because nevirapine induces its own metabolism with multiple dosing, this single dose study may not reflect the impact of hepatic impairment on multiple dose pharmacokinetics (see Section 4.4 Special Warnings and Precautions for Use).
In a 200 mg nevirapine single dose pharmacokinetic study of HIV negative patients with mild and moderate hepatic impairment (Child-Pugh A, n = 6; Child-Pugh B, n = 4), a significant increase in the AUC of nevirapine was observed in one Child-Pugh B patient with ascites suggesting that patients with worsening hepatic function and ascites may be at risk of accumulating nevirapine in the systemic circulation. Because nevirapine induces its own metabolism with multiple dosing, this single dose study may not reflect the impact of hepatic impairment on multiple dose pharmacokinetics (see Section 4.4 Special Warnings and Precautions for Use). Chemical name: 11-cyclopropyl-5, 11-dihydro-4-methyl- 6H-dipyrido[3,2-b:2',3'-e] [1,4]diazepin-6-one.
Chemical name: 11-cyclopropyl-5, 11-dihydro-4-methyl- 6H-dipyrido[3,2-b:2',3'-e] [1,4]diazepin-6-one.