1 Name of Medicine
Avalglucosidase alfa.
2 Qualitative and Quantitative Composition
Each single-use vial contains 100 mg of avalglucosidase alfa* as powder for injection. The powder for injection is reconstituted with 10 mL sterile water for injections (WFI).
Each vial contains an overfill to compensate for liquid loss during preparation.
Following reconstitution, each vial contains 10.3 mL reconstituted solution and a total extractable volume of 10 mL. Each mL of the reconstituted solution contains 10 mg of avalglucosidase alfa.
* Avalglucosidase alfa is a human acid α-glucosidase produced in Chinese hamster ovary cells (CHO) by recombinant DNA technology, which is subsequently conjugated with approximately 7 hexamannose structures (each containing two terminal mannose-6-phosphate (M6P) moieties) to oxidised sialic acid residues on the molecule, thereby increasing bis-M6P levels.
For the full list of excipients, see Section 6.1.
3 Pharmaceutical Form
Powder for injection.
White to pale yellow lyophilised powder.
4.1 Therapeutic Indications
Nexviazyme is indicated for long-term enzyme replacement therapy for the treatment of patients one year of age and older with Pompe disease (acid α-glucosidase deficiency).
4.2 Dose and Method of Administration
Nexviazyme treatment should be supervised by a physician experienced in the management of patients with Pompe disease or other inherited metabolic or neuromuscular diseases.
Dose.
The recommended dose of Nexviazyme is 20 mg/kg of body weight administered every other week as an intravenous infusion. Dose escalation to 40 mg/kg every other week may be considered for patients with infantile onset Pompe disease (IOPD) who experience insufficient control or declining response at the lower dose (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Method of administration.
Preparation for intravenous infusion.
Use aseptic technique during preparation.
1. Determine the number of vials to be reconstituted based on individual patient's weight and the recommended dose of 20 mg/kg or 40 mg/kg.
Patient weight (kg) x dose (mg/kg) = patient dose (in mg).
Patient dose (in mg) divided by 100 mg/vial = number of vials to reconstitute.
If the number of vials includes a fraction, round up to the next whole number.
See Table 1.
 2. Remove the required number of vials needed for the infusion from the refrigerator and set aside for approximately 30 minutes to allow them to reach room temperature.
2. Remove the required number of vials needed for the infusion from the refrigerator and set aside for approximately 30 minutes to allow them to reach room temperature.
3. Reconstitute each vial by slowly injecting 10 mL of Sterile Water for Injections (WFI) to each vial. Each vial will yield 100 mg/10 mL (10 mg/mL). Avoid forceful impact of the water for injection on the powder and avoid foaming. This is performed by slow drop-wise addition of the WFI down the inside of the vial and not directly onto the lyophilised cake. Tilt and roll each vial gently. Do not invert, swirl, or shake. Avoid any air introduction into the infusion bag during the dilution of the product.
4. Perform an immediate visual inspection on the reconstituted vials for particulate matter and discolouration. If upon immediate inspection particles are observed or if the solution is discoloured, do not use. Allow the solution to become dissolved.
5. The reconstituted solution should be diluted in 5% dextrose in water to a final concentration of 0.5 mg/mL to 4 mg/mL. See Table 2 for the recommended total infusion volume based on the patient weight.
6. Slowly withdraw the volume of reconstituted solution from each vial (calculated according to patient's weight).
7. Add the reconstituted solution slowly and directly into the 5% dextrose solution. Avoid foaming or agitation of the infusion bag. Avoid air introduction into the infusion bag.
8. Gently invert or massage the infusion bag to mix. Do not shake.
9. It is recommended to use an in-line, low protein binding, 0.2 microM filter to administer Nexviazyme. After the infusion is complete, flush with dextrose 5% in water bag.
10. Do not infuse Nexviazyme in the same intravenous line with other products.

Administration.
Nexviazyme should be administered as an intravenous infusion. Infusion should be administered incrementally as determined by patient response and comfort. It is recommended that the infusion begins at an initial rate of 1 mg/kg/hour and is gradually increased every 30 minutes if there are no signs of infusion-associated reactions (IARs) in accordance with Table 3. Vital signs should be obtained at each step, before increasing the infusion rate. Patients may be pre-treated with antihistamines, antipyretics and/or corticosteroids to prevent or reduce allergic reactions.
 In the event of anaphylaxis or severe hypersensitivity reaction or severe infusion associated reactions (IARs), immediately discontinue administration of Nexviazyme and initiate appropriate medical treatment. In the event of mild to moderate hypersensitivity reactions or IARs, the infusion rate may be slowed or temporarily stopped and/or appropriate medical treatment initiated (see Section 4.4).
In the event of anaphylaxis or severe hypersensitivity reaction or severe infusion associated reactions (IARs), immediately discontinue administration of Nexviazyme and initiate appropriate medical treatment. In the event of mild to moderate hypersensitivity reactions or IARs, the infusion rate may be slowed or temporarily stopped and/or appropriate medical treatment initiated (see Section 4.4).
Symptoms may persist despite temporarily stopping the infusion; therefore, the treating physician should wait at least 30 minutes for symptoms of the reactions to resolve before deciding to stop the infusion for the remainder of the day. If symptoms subside, resume infusion rate for 30 minutes at half the rate, or less, of the rate at which the reactions occurred, followed by an increase in infusion rate by 50% for 15 to 30 minutes. If symptoms do not recur, increase the infusion rate to the rate at which the reactions occurred and consider continuing to increase the rate in a stepwise manner until the maximum rate is achieved.
Nexviazyme is for single use in one patient only. Contains no antimicrobial preservative.
Special populations.
Paediatric population.
The safety and efficacy of avalglucosidase alfa were assessed in 22 patients with IOPD (1 to 12 years of age) and 2 paediatric patients with LOPD (9 and 16 years of age) in 2 different clinical studies (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties, Clinical trials). There are no data available in patients younger than one year.
Elderly patients.
Clinical studies with Nexviazyme included 14 patients aged 65-75 years and 3 patients over the age of 75 years. There is no recommended dose adjustment for patients over the age of 65 (see Section 4.8).
Hepatic impairment.
The safety and efficacy of Nexviazyme have not been studied in patients with hepatic impairment.
Renal impairment.
No dose adjustment is required in patients with mild renal impairment. Nexviazyme has not been studied in patients with moderate or severe renal impairment (see Section 5.2).
Home infusion.
Home administration by a trained health care professional may be considered for individual patients after safety and tolerability has been established in the clinical setting.4.3 Contraindications
Life-threatening hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 when re-challenge was unsuccessful (see Section 4.4).
4.4 Special Warnings and Precautions for Use
Hypersensitivity reactions including anaphylaxis.
Hypersensitivity reactions, including anaphylaxis, have been reported in Nexviazyme treated patients. In clinical studies 86 (60.6%) patients experienced hypersensitivity reactions including 7 patients who reported severe hypersensitivity reactions and 4 patients who experienced anaphylaxis. Some of the hypersensitivity reactions were IgE mediated. Anaphylaxis signs and symptoms included tongue oedema, hypotension, hypoxia, respiratory distress, chest discomfort, cough, breath sounds abnormal, oxygen saturation decreased, throat tightness, dysphagia, nausea, lip swelling, swollen tongue, dysarthria, dizziness, generalised oedema, flushing, feeling hot, erythema, palmar erythema, and pruritus. Symptoms of severe hypersensitivity reactions included tongue oedema, respiratory failure, respiratory distress, generalised oedema, erythema, urticaria and rash.
Appropriate medical support measures, including cardiopulmonary resuscitation equipment especially for patients with cardiac hypertrophy and patients with significantly compromised respiratory function, should be readily available when Nexviazyme is administered.
If severe hypersensitivity or anaphylaxis occur, Nexviazyme should be discontinued immediately, and appropriate medical treatment should be initiated. The risks and benefits of re-administering Nexviazyme following anaphylaxis or severe hypersensitivity reaction should be considered. Some patients have been re-challenged using slower infusion rates at a dose lower than the recommended dose. In patients with severe hypersensitivity, desensitisation procedure to Nexviazyme may be considered. If the decision is made to re-administer the product, extreme caution should be exercised, with appropriate resuscitation measures available. Once a patient tolerates the infusion, the dose may be increased to reach the approved dose.
If mild or moderate hypersensitivity reactions occur, the infusion rate may be slowed or temporarily stopped.
Infusion associated reactions.
In clinical studies, IARs were reported to occur at any time during and/or within a few hours after the infusion of Nexviazyme and were more likely with higher infusion rates. IARs were reported in approximately 39.4% of patients treated with Nexviazyme in clinical studies. The majority of IARs were assessed as mild to moderate and symptoms reported in more than one patient included respiratory distress, chest discomfort, dyspnoea, cough, oxygen saturation decreased, throat irritation, dyspepsia, nausea, vomiting, diarrhoea, lip swelling, erythema, palmar erythema, rash, rash erythematous, pruritus, urticaria, hyperhidrosis, skin plaque, ocular hyperemia, eyelid oedema, face oedema, increased or decreased blood pressure, tachycardia, headache, dizziness, tremor, burning sensation, pain (including pain in extremity, abdominal pain upper, oropharyngeal pain, and flank pain) somnolence, sluggishness, fatigue, pyrexia, influenza like illness, chills, flushing, feeling hot or cold, cyanosis, and pallor (see Section 4.8 Adverse Effects (Undesirable Effects)). In clinical studies, 6 (4.2%) patients reported severe IARs including symptoms of respiratory distress, hypoxia, chest discomfort, generalised oedema, tongue oedema, dysphagia, nausea, erythema, urticaria, and increased or decreased blood pressure.
Patients with an acute underlying illness at the time of Nexviazyme infusion appear to be at greater risk for IARs. Patients with advanced Pompe disease may have compromised cardiac and respiratory function, which may predispose them to a higher risk of severe complications from IARs. Antihistamines, antipyretics, and/or corticosteroids can be given to prevent or reduce IARs. However, IARs may still occur in patients after receiving pretreatment.
If severe IARs occur, immediate discontinuation of the administration of Nexviazyme should be considered and appropriate medical treatment should be initiated. The benefits and risks of re-administering Nexviazyme following severe IARs should be considered. Some patients have been re-challenged using slower infusion rates at a dose lower than the recommended dose. Once a patient tolerates the infusion, the dose may be increased to reach the approved dose. If a mild or moderate IARs occur regardless of pre-treatment, decreasing the infusion rate or temporarily stopping the infusion may ameliorate the symptoms (see Section 4.8).
Immunogenicity.
Treatment emergent anti-drug antibodies (ADA) were reported in both treatment naïve (95%) and treatment experienced patients (62%) (see Section 4.8).
IARs and hypersensitivity reactions may occur independent of the development of ADA. The majority of IARs and hypersensitivity reactions were mild or moderate and were managed with standard clinical practices. In treatment-naïve patients, a trend for increases in the incidence of IARs was observed with increasing ADA titres, with the highest incidence of IARs (69.2%) reported in the high ADA peak titre range ≥ 12,800, compared with an incidence of 33.3% in patients with intermediate ADA titre 1,600-6,400, an incidence of 14.3% in those with low ADA titre 100-800 and an incidence of 33.3% in those who were ADA negative (33.3%). In clinical studies, there was no apparent clinically significant effect of ADA on efficacy in most patients (see Section 4.8).
ADA testing may be considered if patients do not respond to therapy. Adverse-event-driven immunologic testing, including IgG and IgE ADA, may be considered for patients who have risk for allergic reaction or previous anaphylactic reaction to alglucosidase alfa.
If testing is warranted, contact Sanofi Genzyme at 1800 818 806 for information on Sanofi Genzyme's Rare Disease Specialty Testing Program.
Risk of acute cardiorespiratory failure.
Caution should be exercised when administering Nexviazyme to patients susceptible to fluid volume overload or patients with acute underlying respiratory illness or compromised cardiac and/or respiratory function for whom fluid restriction is indicated. These patients may be at risk of serious exacerbation of their cardiac or respiratory status during infusion. Appropriate medical support and monitoring measures should be readily available during Nexviazyme infusion, and some patients may require prolonged observation times that should be based on the individual needs of the patient.
Cardiac arrhythmia and sudden death during general anaesthesia for central venous catheter placement.
Caution should be used when administering general anaesthesia for the placement of a central venous catheter or for other surgical procedures in patients with IOPD with cardiac hypertrophy. Cardiac arrhythmia, including ventricular fibrillation, ventricular tachycardia, and bradycardia, resulting in cardiac arrest or death, or requiring cardiac resuscitation or defibrillation, have been associated with the use of general anaesthesia in IOPD patients with cardiac hypertrophy.
Use in renal impairment.
No dose adjustment is required in patients with mild renal impairment. Nexviazyme has not been studied in patients with moderate or severe renal impairment (see Section 5.2).
Use in the elderly.
Clinical studies with Nexviazyme included 14 patients aged 65-75 years and 3 patients over the age of 75 years. There is no recommended dose adjustment for patients over the age of 65 (see Section 4.8).
Hepatic impairment.
The safety and efficacy of Nexviazyme have not been studied in patients with hepatic impairment.
Paediatric use.
The safety and efficacy of avalglucosidase alfa were assessed in 22 patients with IOPD (1 to 12 years of age) and 2 paediatric patients with LOPD (9 and 16 years of age) in 2 different clinical studies (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties, Clinical trials). There are no data available in patients younger than one year.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no clinical data on the effects of Nexviazyme on human fertility. Avalglucosidase alfa caused no adverse effects in a combined male and female fertility study in mice up to 50 mg/kg IV every other day.
(Category B1)
There are no available data on the use of Nexviazyme in pregnant women. In an embryofetal toxicity study in mice, administration of avalglucosidase alfa during the period of organogenesis produced maternal toxicity related to an immunological response (including an anaphylactoid response) at the highest dose of 50 mg/kg/day (17 times the human steady-state AUC at the recommended biweekly dose of 20 mg/kg for patients with LOPD). This dose also produced increased fetal loss. Avalglucosidase alfa does not cross the placenta in mice, suggesting that the embryofetal effects were related to maternal toxicity from the immunological response. No malformations or developmental variations were observed. The developmental no-observed-adverse-effect level (NOAEL) in mice was 20 mg/kg/day (4.8 times the human steady-state AUC at the recommended biweekly dose of 20 mg/kg for patients with LOPD). No adverse effects were observed in an embryofetal toxicity study in rabbits administered avalglucosidase alfa during the period of organogenesis up to 100 mg/kg/day IV (91 times the human steady-state AUC at the recommended biweekly dose of 20 mg/kg for patients with LOPD).
The potential risk for humans is unknown. No conclusions can be drawn regarding whether or not Nexviazyme is safe for use during pregnancy. Nexviazyme should be used during pregnancy only if the potential benefits to the mother outweigh the potential risks, including those to the fetus.
There are no available data on the presence of Nexviazyme in human milk or the effects of Nexviazyme on milk production or the breastfed infant. No conclusions can be drawn regarding whether or not Nexviazyme is safe for use during breastfeeding. Nexviazyme should be used during breastfeeding only if the potential benefits to the mother outweigh the potential risks, including those to the breastfed child.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed. Because dizziness, hypotension and fatigue have been reported as IARs, this may affect the ability to drive and use machines on the day of the infusion (see Section 4.8).
4.8 Adverse Effects (Undesirable Effects)
Adverse drug reactions per System Organ Class are presented using the following CIOMS frequency rating: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000) and not known (cannot be estimated from the available data). Table 4 includes common ADRs reported in patients treated with Nexviazyme in the pooled analysis of clinical studies (142 patients) and additionally includes treatment-related adverse events that are considered biologically plausibly related to Nexviazyme based on ADRs reported with alglucosidase alfa treatment.
Due to the small patient population, an adverse drug reaction reported in 2 patients is classified as common.
Summary of the safety profile.
The pooled safety analysis from 4 clinical studies EFC14028 (COMET), ACT14132 (mini-COMET), TDR12857 (NEO), and LTS13769 (NEO-EXT) included a total of 142 patients (118 adult and 24 paediatric patients (1 paediatric patient directly enrolled in the open-label extension period of Study 1)) treated with Nexviazyme.
Serious adverse drug reactions reported in patients treated with Nexviazyme were respiratory distress (1.4%), chills (1.4%), headache (0.7%), dyspnoea (0.7%), hypoxia (0.7%), tongue oedema (0.7%), nausea (0.7%), pruritus (0.7%), urticaria (0.7%), skin discoloration (0.7%), chest discomfort (0.7%), pyrexia (0.7%), blood pressure increased (0.7%), hypotension (0.7%), body temperature increased (0.7%), heart rate increase (0.7%), and oxygen saturation decreased (0.7%). A total of 4 patients receiving Nexviazyme in clinical studies permanently discontinued treatment due to adverse drug reactions, including 3 patients who discontinued the treatment because of a serious adverse drug reaction. The most frequently reported adverse drug reactions (ADRs) (> 5%) were pruritus, nausea, headache, rash, urticaria, chills, fatigue, and erythema. IARs were reported in 59 (39.4%) patients. IARs reported in more than 1 patient included respiratory distress, chest discomfort, dyspnoea, cough, oxygen saturation decreased, erythema, palmar erythema, rash, rash erythematous, pruritus, urticaria, skin plaque, throat irritation, dyspepsia, nausea, vomiting, diarrhoea, lip swelling, swollen tongue, ocular hyperemia, eyelid oedema, face oedema, increased blood pressure, hypotension, tachycardia, headache, dizziness, tremor, burning sensation, pain (including pain in extremity, abdominal pain upper, oropharyngeal pain, and flank pain), somnolence, sluggishness, fatigue, pyrexia, influenza-like illness, chills, flushing, feeling hot or cold, hyperhidrosis, cyanosis, and pallor. The majority of IARs were assessed as mild to moderate (see Section 4.4).
Common ADRs (ADRs occurring in greater than or equal to 1% of patients) reported in patients treated with Nexviazyme in the pooled analysis of clinical studies (142 patients) are listed in Table 4. Table 4 additionally includes treatment-related adverse events that are considered biologically plausibly related to Nexviazyme based on ADRs reported with alglucosidase alfa treatment.
 In a comparative study, EFC14028 (COMET), 100 LOPD patients aged 16 to 78 naïve to enzyme replacement therapy were treated every other week either with 20 mg/kg of Nexviazyme (n = 51) or 20 mg/kg of alglucosidase alfa (n = 49). During the double-blind active-controlled period of 49 weeks, serious adverse drug reactions were reported in 2% of patients treated with Nexviazyme and 6.1% of those treated with alglucosidase alfa. A total of 4 patients receiving alglucosidase alfa in the study permanently discontinued treatment due to adverse drug reactions; none of the patients from the Nexviazyme group permanently discontinued the treatment. The most frequently reported ADRs (> 5%) in patients treated with Nexviazyme were headache, nausea, pruritus, urticaria, and fatigue.
In a comparative study, EFC14028 (COMET), 100 LOPD patients aged 16 to 78 naïve to enzyme replacement therapy were treated every other week either with 20 mg/kg of Nexviazyme (n = 51) or 20 mg/kg of alglucosidase alfa (n = 49). During the double-blind active-controlled period of 49 weeks, serious adverse drug reactions were reported in 2% of patients treated with Nexviazyme and 6.1% of those treated with alglucosidase alfa. A total of 4 patients receiving alglucosidase alfa in the study permanently discontinued treatment due to adverse drug reactions; none of the patients from the Nexviazyme group permanently discontinued the treatment. The most frequently reported ADRs (> 5%) in patients treated with Nexviazyme were headache, nausea, pruritus, urticaria, and fatigue.
The most commonly reported treatment-emergent adverse events (TEAEs) (≥ 20%) were nasopharyngitis (23.5%), back pain (23.5%), and headache (21.6%) in the Nexviazyme group and headache (32.7%), nasopharyngitis (24.5%), and fall (20.4%) in the alglucosidase alfa group. See Table 5.
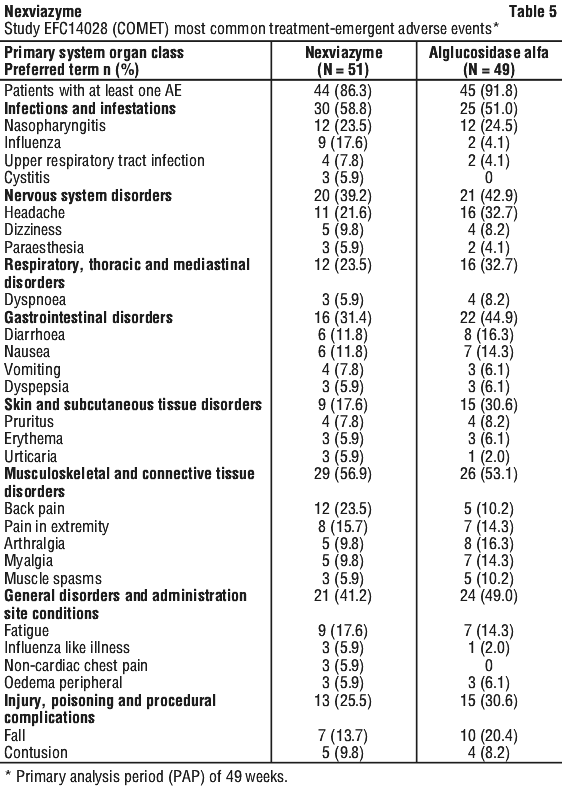 IARs were reported in 25.5% of the patients treated with Nexviazyme, compared to 32.7% of patients treated with alglucosidase alfa. The most frequently reported treatment-emergent IARs (> 2 patients) in the Nexviazyme group were pruritus and urticaria, and in the alglucosidase alfa group were nausea, pruritus, and flushing. All the IARs reported in more than 1 patient were mild to moderate and included headache, chills, dizziness, dyspnoea, erythema, flushing, diarrhoea, nausea, pruritus, rash, urticaria and feeling hot. Severe IARs were reported in 2 patients treated with alglucosidase alfa; there were no reports of severe IARs in patients treated with Nexviazyme.
IARs were reported in 25.5% of the patients treated with Nexviazyme, compared to 32.7% of patients treated with alglucosidase alfa. The most frequently reported treatment-emergent IARs (> 2 patients) in the Nexviazyme group were pruritus and urticaria, and in the alglucosidase alfa group were nausea, pruritus, and flushing. All the IARs reported in more than 1 patient were mild to moderate and included headache, chills, dizziness, dyspnoea, erythema, flushing, diarrhoea, nausea, pruritus, rash, urticaria and feeling hot. Severe IARs were reported in 2 patients treated with alglucosidase alfa; there were no reports of severe IARs in patients treated with Nexviazyme.
The 95 patients who entered open-label extension period of EFC14028 (COMET) consisted of 51 patients who continued treatment with Nexviazyme and 44 patients who switched from alglucosidase alfa to Nexviazyme. During the open-label extension period serious adverse drug reactions were reported by 3 (5.8%) patients continuing Nexviazyme treatment throughout the study and by 3 (6.8%) patients who switched to Nexviazyme. The most frequently reported adverse drug reactions (> 5%) by patients continuing Nexviazyme treatment throughout the study were nausea, chills, erythema, pruritus, and urticaria. The most frequently reported adverse drug reactions (> 5%) by patients who switched to Nexviazyme were pruritus, rash, headache, nausea, chills, fatigue, and urticaria.
IARs were reported in 12 (23.5%) patients continuing Nexviazyme treatment throughout the study; IARs reported in more than 1 patient were nausea, chills, pyrexia, erythema, pruritus, urticaria, rash, and ocular hyperemia. IARs were reported in 22 (50%) patients who switched to Nexviazyme; IARs reported in more than 1 patient were pruritus, headache, rash, nausea, chills, fatigue, urticaria, respiratory distress, feeling cold, chest discomfort, erythema, rash erythematous, rash pruritic, skin plaque, lip swelling, swollen tongue and burning sensation. The number of IARs in both groups decreased over time.
No adverse drug reaction or IAR was reported by the additional paediatric patient directly enrolled in the open-label extension period.
Description of selected adverse drug reactions.
Immunogenicity.
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralising antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Nexviazyme in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
The incidence of ADA response to avalglucosidase alfa in Nexviazyme-treated patients with Pompe disease is shown in Table 6. The median time to seroconversion was 8.3 weeks.
In treatment-naïve adult patients, the occurrence of IAR was observed in both ADA-positive and ADA-negative patients. Increase in the incidence of IAR and hypersensitivity were observed with higher IgG ADA titres. In enzyme replacement therapy (ERT) experienced adult patients, the occurrences of IARs and hypersensitivity were higher in patients who developed treatment emergent ADA compared to patients who were ADA negative. One treatment naïve patient and two treatment experienced patients developed anaphylaxis. The occurrences of IARs were similar between paediatric patients with ADA positive and negative status. One treatment-experienced paediatric patient developed anaphylaxis (see Section 4.4).
In clinical study EFC14028 (COMET), 81 of 96 (84.4%) patients developed treatment-emergent ADA. Majority of patients developed ADA titres in the low to intermediate range, with 7 patients reported having High Sustained Antibody Titres (HSAT) to Nexviazyme. Evaluation of ADA cross reactivity at week 49 showed that patients generate antibodies that are cross-reactive to alglucosidase alfa and antibodies specific to Nexviazyme were detected in 3 (5.9%) patients. Variable impacts on PK, PD and efficacy measures were observed among high titre patients, however, in most patients there was no apparent clinically significant effect of ADA on efficacy (see Section 5.2).

Paediatric population.
Adverse drug reactions reported from clinical trials in the paediatric population (19 paediatric patients with IOPD and 1 paediatric patient with LOPD) were similar to those reported in adults.
In Study ACT14132 (mini-COMET) in IOPD patients, which compared avalglucosidase alfa with alglucosidase alfa, 5/5 patients (100%) in the avalglucosidase alfa group reported adverse events compared with 5/6 patients (83%) in the alglucosidase group. See Table 7.

Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
IARs are more likely to occur with higher infusion rates (see Section 4.4). In a clinical study, paediatric patients received doses up to 40 mg/kg of body weight. For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Alimentary tract and metabolism products, enzymes.
ATC code: A16AB22.
Mechanism of action.
Pompe disease (also known as glycogen storage disease type II, acid maltase deficiency, and glycogenosis type II) is a rare metabolic muscle disease inherited in an autosomal recessive manner defined by a deficiency of acid α-glucosidase (GAA), which is necessary for the degradation of lysosomal glycogen. GAA cleaves alfa-1,4 and alfa-1,6 linkages in glycogen under the acidic conditions of the lysosome. Pompe disease results in intra-lysosomal accumulation of glycogen in various tissues, particularly cardiac and skeletal muscles, leading to the development of cardiomyopathy, progressive muscle weakness, and impairment of respiratory function.
Avalglucosidase alfa is a recombinant human acid α-glucosidase (rhGAA) that provides an exogenous source of GAA. Avalglucosidase alfa is a modification of alglucosidase alfa in which approximately 7 hexamannose structures each containing 2 terminal mannose-6-phosphate (bis-M6P) moieties are conjugated to oxidised sialic acid residues on alglucosidase alfa. Avalglucosidase alfa has a 15-fold increase in mannose-6-phosphate (M6P) moieties compared with alglucosidase alfa. Increasing the level of bis-M6P on rhGAA provides a mechanism to drive uptake into the diaphragm and other skeletal muscle via the cation-independent M6P receptor, where it can degrade glycogen and ameliorate tissue damage.
Pharmacodynamic effects.
In treatment-naïve LOPD patients aged 16 to 78, the mean percentage (SD) change in urinary hexose tetrasaccharides from baseline for patients treated with Nexviazyme 20 mg/kg every other week and alglucosidase alfa 20 mg/kg every other week was -53.90% (24.03) and -10.8% (32.33), respectively, week 49.
In paediatric IOPD patients (< 18 years of age) treated with Nexviazyme at 40 mg/kg every other week who demonstrated either clinical decline (cohort 2) or sub-optimal clinical response (cohort 3) while on treatment with alglucosidase alfa, the mean percentage (SD) change in urinary hexose tetrasaccharides from baseline was -40.97% (16.72) and -37.48% (17.16), respectively, after 6 months. In patients previously declining treatment with Nexviazyme at 20 mg/kg every other week, the mean (SD) percentage change was 0.34% (42.09).
Clinical trials.
The safety and efficacy of Nexviazyme have been evaluated in clinical studies of patients who were either naïve or treatment experienced at the initiation of treatment.
Clinical trials in patients with LOPD.
Study EFC14028 (COMET), was a multinational, multi-centre, randomised, double-blinded study comparing the efficacy and safety of Nexviazyme and alglucosidase alfa in 100 treatment-naïve LOPD patients aged 16 to 78 years at the initiation of treatment. Patients were randomised in a 1:1 ratio based on baseline forced vital capacity (FVC), gender, age, and country to receive 20 mg/kg of Nexviazyme or alglucosidase alfa once every other week for 12 months (49 weeks).
Study EFC14028 (COMET) included an open-label extension treatment period where all patients in the alglucosidase arm were switched to Nexviazyme and continued treatment up to at least week 145. Overall, 95 patients entered the open-label period (51 from the Nexviazyme arm and 44 from the alglucosidase arm). An additional paediatric patient was enrolled directly into the extension treatment period with Nexviazyme.
The primary endpoint of study EFC14028 (COMET) was the change in FVC (% predicted) in the upright position from baseline to 12 months (week 49). At week 49, the least square (LS) mean change (SE) in FVC % predicted for patients treated with Nexviazyme and alglucosidase alfa was 2.89% (0.88) and 0.46% (0.93), respectively.
The clinically significant LS mean difference of 2.43% (95% CI: -0.13, 4.99) between Nexviazyme and alglucosidase alfa FVC % predicted exceeded the pre-defined non-inferiority margin of -1.1 and achieved statistical non-inferiority (p = 0.0074). The study did not demonstrate statistical significance for superiority (p = 0.0626) and the testing of the secondary endpoints was performed without multiplicity adjustment.
The results for the primary endpoint are detailed in Table 8 and Figure 1.

 For patients who switched from alglucosidase to Nexviazyme treatment after week 49, the LS mean change in FVC % predicted from week 49 to week 145 was 0.7 (1.1) (95% CI: -1.4, 2.8). A stabilisation of FVC % predicted was observed after the switch to Nexviazyme in the alglucosidase alfa group with similar values to the Nexviazyme group at week 145. Patients who continued in the Nexviazyme arm maintained an improvement in FVC % predicted compared with baseline.
For patients who switched from alglucosidase to Nexviazyme treatment after week 49, the LS mean change in FVC % predicted from week 49 to week 145 was 0.7 (1.1) (95% CI: -1.4, 2.8). A stabilisation of FVC % predicted was observed after the switch to Nexviazyme in the alglucosidase alfa group with similar values to the Nexviazyme group at week 145. Patients who continued in the Nexviazyme arm maintained an improvement in FVC % predicted compared with baseline.
The key secondary endpoint of study EFC14028 (COMET) was change in total distance walked in 6 minutes (6-Minute Walk Test, 6MWT) from baseline to 12 months (week 49). At week 49, the LS mean change from baseline (SE) in 6MWT for patients treated with Nexviazyme and alglucosidase alfa was 32.21 m (9.93) and 2.19 m (10.40) respectively. The LS mean difference of 30.01 m (95% CI: 1.33, 58.69) showed numerical improvement with Nexviazyme compared with alglucosidase alfa. The results for the 6MWT are detailed in Table 9 and Figure 2.

 For patients who switched from alglucosidase alfa to Nexviazyme treatment after week 49, the LS mean change in 6MWT (distance walked in metres) from week 49 to week 145 was -2.3 (10.6), 95% CI: -23.2, 18.7. At Week 145, a stabilisation in 6MWT was observed after the switch from the alglucosidase alfa group to Nexviazyme. The Nexviazyme arm participants sustained the improvement compared with baseline.
For patients who switched from alglucosidase alfa to Nexviazyme treatment after week 49, the LS mean change in 6MWT (distance walked in metres) from week 49 to week 145 was -2.3 (10.6), 95% CI: -23.2, 18.7. At Week 145, a stabilisation in 6MWT was observed after the switch from the alglucosidase alfa group to Nexviazyme. The Nexviazyme arm participants sustained the improvement compared with baseline.
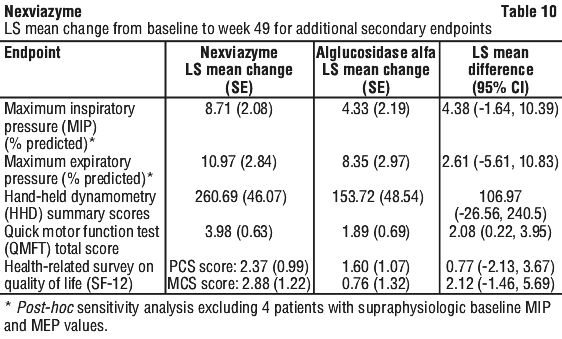
Additional secondary endpoints of the study were maximum inspiratory pressure (MIP), maximum expiratory pressure (MEP), Hand-held dynamometry (HHD) summary score, quick motor function test (QMFT) total score, and SF-12 (health-related survey on quality of life, both physical and mental component scores). The results for these endpoints are detailed in Table 10.
Clinical trial in patients with IOPD.
Study ACT14132 (mini-COMET) was a multi-stage, phase 2, open-label, multi-centre, multinational, repeated ascending dose cohort of Nexviazyme in paediatric IOPD patients 1 to 12 years of age who demonstrated either clinical decline or sub-optimal clinical response while on treatment with alglucosidase alfa. The study enrolled a total of 22 patients; cohort 1 had 6 patients who demonstrated clinical decline and received 20 mg/kg every other week for 25 weeks, cohort 2 had 5 patients who demonstrated clinical decline and received 40 mg/kg every other week for 25 weeks, and cohort 3 had 11 patients who demonstrated sub-optimal response and received either Nexviazyme at 40 mg/kg every other week for 25 weeks (5 patients) or alglucosidase alfa at their stable pre-study dose (ranging between 20 mg/kg every other week and 40 mg/kg weekly) for 25 weeks (6 patients).
The primary endpoint of study ACT14132 (mini-COMET) was to evaluate the safety and tolerability of administering Nexviazyme. The secondary endpoint was to determine the efficacy of Nexviazyme. Data showed stabilisation or improvement in efficacy outcomes of Gross Motor Function Measure (GMFM-88), Quick Motor Function Test (QMFT), Pompe-Paediatric Evaluation of Disability Inventory (Pompe-PEDI), Left Ventricular Mass Z (LVMZ) score, eyelid position measurements in patients previously declining or insufficiently controlled with alglucosidase alfa. Treatment effect was more pronounced with 40 mg/kg every other week compared to the 20 mg/kg every other week. Two out of six patients treated with Nexviazyme at 20 mg/kg every other week (cohort 1) demonstrated further clinical decline and received dose increase from 20 to 40 mg/kg every other week at week 55 and 61 respectively. All patients who received 40 mg/kg every other week maintained this dose for the duration of the study.
The long-term effects of treatment with Nexviazyme were evaluated in 10 patients at week 49, 8 patients at week 73, and 3 patients at week 97. In patients with IOPD previously declining with alglucosidase alfa, the efficacy on specific parameters of decline, including motor function, cardiac left ventricular mass, and eyelid position measurements, was sustained up to 2 years.
5.2 Pharmacokinetic Properties
Patients with late-onset Pompe disease (LOPD).
The pharmacokinetics of avalglucosidase alfa was evaluated in a population analysis of 75 LOPD patients aged 16 to 78 years who received 5 to 20 mg/kg of avalglucosidase alfa every other week for up to 5 years.
Patients with infantile-onset Pompe disease (IOPD).
The pharmacokinetics of avalglucosidase alfa was characterised in 16 patients aged 1 to 12 years who were treated with avalglucosidase alfa, which included 6 patients treated with 20 mg/kg and 10 patients treated with 40 mg/kg doses every other week for up to 25 weeks.
Absorption.
In LOPD patients, for a 4-hour IV infusion of 20 mg/kg every other week, the mean Cmax (CV%) and mean AUC2W (CV%) were 273 microgram/mL (24%) and 1220 microgram.h/mL (29%), respectively.
In IOPD patients, for a 4-hour IV infusion of 20 mg/kg every other week and 7-hour IV infusion for 40 mg/kg every other week, the mean Cmax ranged from 175 to 189 microgram/mL for the 20 mg/kg dose and 205 to 403 microgram/mL for 40 mg/kg dose. The mean AUC2W ranged from 805 to 923 microgram.hr/mL for the 20 mg/kg dose and 1720 to 2630 microgram.hr/mL for 40 mg/kg dose.
Distribution.
In LOPD patients, the typical population PK model predicted central compartment volume of distribution of avalglucosidase alfa was 3.4 L.
In IOPD patients treated with avalglucosidase alfa 20 mg/kg and 40 mg/kg every other week, the mean volume of distribution at steady state ranged between 3.5 to 5.4 L.
Metabolism.
The metabolic pathway of avalglucosidase alfa has not been characterised. As a glycoprotein, avalglucosidase alfa is expected to be degraded into small peptides or amino acids via non-saturable catabolic pathways.
Excretion.
In LOPD patients, the typical population PK model predicted linear clearance was 0.87 L/h. Following 20 mg/kg every other week, the mean plasma elimination half-life was 1.55 hours.
In IOPD patients treated with avalglucosidase alfa 20 mg/kg and 40 mg/kg every other week, mean plasma clearance ranged from 0.53 to 0.70 L/h, and mean plasma elimination half-life from 0.60 to 1.19 hours.
Immunogenicity.
In the study EFC14028/COMET, 95.2% (59 of 62 patients) receiving Nexviazyme developed treatment-emergent ADA. As only 2 patients were ADA negative, therefore, the ADA impact on PK was assessed by categorising the ADA-positive patients into 3 peak titre groups: ≤ 800, 1,600-6,400, and ≥ 12,800. Five patients had ≥ 50% change in the AUC at week 49 from baseline but no obvious pattern in titres. Inter-subject comparison of the AUC at Day 1 or 2 and week 49 supported the overall analysis of percent change in the AUC and ADA positivity categorized by ADA titres. In vitro evaluation of neutralising antibodies that inhibited enzyme activity or inhibited cellular uptake demonstrated no clear relationship of assay positivity with AUC (see Section 4.8). The treatment-experienced IOPD patients had titres ≤ 6,400, and as changes in PK were not observed, the relationship to ADA was not evaluated for this group.
Special populations.
Population pharmacokinetic analyses in LOPD patients showed that age and gender did not meaningfully influence the pharmacokinetics of avalglucosidase alfa.
Hepatic impairment.
The pharmacokinetics of avalglucosidase alfa has not been studied in patients with hepatic impairment.
Renal impairment.
No formal study of the effect of renal impairment on the pharmacokinetics of avalglucosidase alfa was conducted. On the basis of a population pharmacokinetic analysis of data from 75 LOPD patients receiving 20 mg/kg, including 6 patients with mild renal impairment (glomerular filtration rate: 60 to 89 mL/min; at baseline), no relevant effect of renal impairment on avalglucosidase alfa exposure was observed.
5.3 Preclinical Safety Data
Genotoxicity.
Genotoxic studies have not been conducted with avalglucosidase alfa.
Carcinogenicity.
Carcinogenicity studies have not been conducted with avalglucosidase alfa.6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, histidine hydrochloride monohydrate, glycine, mannitol, polysorbate 80.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store in a refrigerator between 2°C to 8°C. Do not use Nexviazyme after the expiration date on the vial.
The reconstituted and diluted solution should be administered without delay. The reconstituted product can be stored up to 24 hours when refrigerated at 2°C to 8°C and diluted product can be stored up to 24 hours when refrigerated at 2°C to 8°C and up to 9 hours (including infusion time) when stored at room temperature (up to 25°C).
6.5 Nature and Contents of Container
Nexviazyme is supplied in a 20 mL, Type I, colourless, clear, glass vial closed with 20 mm siliconised elastomeric stopper. The stoppered vials are crimped with an aluminium seal with a flip-off button.
Each pack contains 1 vial.
6.6 Special Precautions for Disposal
Remaining Nexviazyme left in a vial after withdrawing the patient's calculated dose should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
CAS number.
1802558-87-7.7 Medicine Schedule (Poisons Standard)
Schedule 4 (Prescription Only Medicine).
Summary Table of Changes
