What is in this leaflet
This leaflet answers some common questions about Norvir.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Norvir is used for
Norvir is used to treat HIV (Human Immunodeficiency Virus) infections.
It belongs to a group of medicines called protease inhibitors.
Norvir works by interfering with the enzyme that the HIV virus needs to infect new cells.
It may be given alone or with certain other anti-HIV medicines. Your doctor will determine which medicines are best for you.
Norvir has not been shown to decrease the chance of transmitting HIV to a sexual partner. You must continue to use safe sexual practices (e.g. condoms) while taking Norvir.
Ask your doctor or pharmacist if you have any questions about why it has been prescribed for you. Your doctor may have prescribed it for another purpose.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children under the age of 12 years.
Before you take Norvir
When you must not take it
Do not take Norvir if you have an allergy to:
- any medicine containing ritonavir
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- hives, rash or itching of the skin.
Do not take Norvir if you are currently taking any of the following medicines:
- alfuzosin hydrochloride (treatment of Benign Prostatic Hyperplasia or BPH in men);
- ranolazine to treat angina;
- amiodarone, bepridil, dronedarone, flecainide, propafenone, quinidine, encainide (medicines to correct irregular heartbeats);
- fusidic acid (an antibiotic);
- apalutamide (treatment for prostate cancer);
- neratinib (treatment for early stage breast cancer);
- venetoclax (treatment for chronic lymphocytic leukaemia);
- voriconazole (used to treat fungal infections);
- colchicine (a treatment for gout) if you have liver or kidney problems;
- astemizole or terfenadine (these antihistamine medicines may be available without prescription);
- rifabutin (used to prevent/treat certain infections);
- blonanserin, clozapine, lurasidone or pimozide (used to treat certain psychological and emotional conditions);
- ergotamine, dihydroergotamine, ergometrine or ethylergometrine (treatment for migraine and headaches);
- cisapride (used to relieve certain stomach problems);
- products that contain St John's Wort (Hypericum perforatum) as this may stop Norvir from working properly. St John's wort is often used in herbal medicines that you can buy yourself;
- lovastatin, simvastain, lomitapide (to lower your cholesterol);
- salmeterol (treatment for asthma);
- piroxicam (used to relieve pain);
- sildenafil if you suffer from a lung disease called pulmonary arterial hypertension that makes breathing difficult. Patients without this disease may use sildenafil for erectile dysfunction under their doctor's supervision;
- pethidine, dextropropoxyphene (used to relieve pain);
- midazolam or triazolam (used to relieve anxiety and/or trouble sleeping);
- clorazepate, diazepam, estazolam, flurazepam, triazolam, midazolam, zolpidem (used to help you sleep and/or relieve anxiety).
Do not take it after the expiry date printed on the bottle or if the packaging is damaged or shows signs of tampering. If it has expired or is damaged return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor or pharmacist if you have any allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor or pharmacist if you have or have had any of the following medical conditions:
- liver problems or problems with your pancreas.
- you have hepatitis B or C, and are being treated with a combination of antiretroviral agents, as you are at a greater risk of developing serious side effects.
- haemophilia, as there have been reports of increased bleeding in patients with haemophilia who are taking this type of medicine.
- diabetes, as there have been reports of worsening of or the development of diabetes in some patients taking this type of medicine.
Tell your doctor or pharmacist if you are pregnant, intend to become pregnant or are breastfeeding. Your doctor or pharmacist can discuss the risks and benefits involved.
If you have not told your doctor or pharmacist about any of the above, tell him or her before you take Norvir.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food store.
If Norvir is taken in combination with other antiviral medicines, it is important that you also carefully read the leaflets that are provided with these other medicines. They may include additional information in those leaflets about situations when Norvir should be avoided.
If you have any further questions about Norvir or the other medicines prescribed, please ask your doctor or pharmacist.
Some medicines and Norvir may interfere with each other. These include:
- didanosine, zidovudine, delaviridine, efavirenz, saquinavir, amprenavir, indinavir, nelfinavir, tipranavir, maraviroc, raltegravir (other antiviral treatments);
- digoxin (used to correct irregular heartbeats);
- abemaciclib, dasatinib, encorafenib, ibrutinib, ivosidenib, neratinib, nilotinib, vincristine, vinblastine (used to treat different types of cancer);
- fostamatinib (used for low platelet count);
- dihydroergotamine, ergotamine (used to treat migraine headache);
- ergometrine, methylergometrine (used to stop excessive bleeding that may occur following childbirth or an abortion);
- buspirone (used to help you sleep and/or relieve anxiety);
- fentanyl (used to relieve pain);
- bedaquiline, delamanid (used to prevent/treat certain infections);
- ketoconazole (used to treat fungal infections);
- clarithromycin, sulphamethasone/trimethoprim, fusidic acid (used to treat infections);
- simeprevir, glecaprevir/pibrentasvir (used to treat chronic hepatitis C);
- warfarin, rivaroxaban (used to thin the blood);
- trazodone (used to treat depression);
- fluticasone, budesonide or triamcinolone (cortisone used to treat inflammation);
- disulfiram (used to treat alcohol dependence);
- metronidazole (used to treat certain parasite infections);
- bupropion (used to help smoking cessation);
- bosentan (used to treat a condition called pulmonary arterial hypertension);
- quetiapine (used to treat psychiatric disorders);
- avanafil, sildenafil, vardenafil (used for erectile dysfunction);
- elagolix (used for moderate to severe pain associated with endometriosis);
- theophylline (used to treat chronic lung conditions).
These medicines may be affected by Norvir or may affect how well it works. You may need to use different amounts of your medicine, or take different medicines.
Your doctor or pharmacist has more information on medicines to be careful with or to avoid while taking Norvir.
Other interactions
There are many other medicines that may not mix with Norvir because their effects could potentially increase or decrease when taken together. In some cases your doctor may need to perform certain tests, change the dose or monitor you regularly.
This is why you should tell your doctor if you are taking any medicines or herbal products, including those you have bought yourself. Your doctor may need to prescribe different amounts of your medicine for you to take, or put you on a different medicine altogether.
Check with your doctor or pharmacist who will have a complete list of medicines which interfere with Norvir.
Norvir affects the way oral contraceptives work.
Take care when taking sildenafil, tadalafil, or vardenafil, used for erectile dysfunction. Norvir interacts with these medicines. The dosage of these medicines should be reduced to avoid damage to the penis. You must not take Norvir with sildenafil if you also suffer from pulmonary arterial hypertension.
How to take Norvir
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take and when to take it
The usual dose is 600mg of ritonavir taken twice a day (6 tablets).
How to take it
Take Norvir with food.
Swallow tablets whole with a full glass of water.
Do not break, crush or chew.
When to take it
Take your Norvir at regular twelve-hour intervals at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take your medicine.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
Norvir is not a cure for your HIV infection, but helps control it. Therefore, Norvir must be taken every day.
Tell your doctor straight away if a side effect is preventing you from taking Norvir.
Always keep enough Norvir on hand so you don't run out. When you travel or need to stay in the hospital, make sure you have enough Norvir to last until you can get a new supply.
Do not stop taking Norvir without talking with your doctor, even if you feel better.
Taking Norvir as recommended should give you the best chance of delaying resistance to the medicines. You may continue to develop infections or other illnesses associated with HIV disease while you are taking Norvir.
If you forget to take it
If you forget to take it and it is almost time for you to take your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose that you missed.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally. This may increase the chance of getting an unwanted side effect.
Ask your doctor or pharmacist if you are not sure what to do.
Ask your pharmacist for hints if you have trouble remembering when to take your medicine.
If you take too much (overdose)
Immediately telephone your doctor, or the Poisons Information Centre (telephone Australia 13 11 26 or New Zealand 0800 764766), or go to Accident and Emergency at your nearest hospital if you think you or anyone else may have taken too much Norvir.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include tingling, prickling, or numbness of the skin.
While you are using Norvir
Things you must do
Tell your doctor or pharmacist immediately if you become pregnant while you are taking this medicine.
Norvir may pass into breast milk. To avoid transmitting the infection, mothers with HIV should not breast feed their babies.
Remind your doctor that you are taking Norvir if you are about to be started on any new medicine.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
Tell your surgeon that you are taking this medicine if you are going to have surgery.
Tell your doctor that you are taking this medicine if you are about to have any blood tests. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not take this medicine to treat any other complaints unless your doctor or pharmacist tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not stop taking Norvir, or change the dosage, without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how Norvir affects you. Norvir generally does not cause problems with your ability to drive a car or operate machinery. However, as with many medicines, Norvir may cause dizziness, sleepiness and nausea in some people.
Make sure you know how you react to Norvir before you drive a car or operate machinery.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Norvir.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Frequently, it is difficult to tell whether side effects are the result of taking Norvir, effects of the HIV disease or side effects of other medicines you may be taking. For this reason, it is very important to inform your doctor of any change in your condition.
Your doctor may want to change your dose or advise you to stop taking Norvir.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- nausea, vomiting
- diarrhoea
- feeling of weakness
- stomach ache
- headache
- tingling sensation
- change in taste sensation
- feeling weak / tired
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
The following side effects have also been reported:
- allergic reactions including skin rashes (may be red, raised, itchy), severe swelling of the skin and other tissues
- difficulty in breathing
- flushing of the skin
- dizziness
- inability to sleep (insomnia)
- anxiety
- sleepiness
- numbness
- unusual sensitivity of the skin
- heartburn
- sore throat
- increased cough
- wind (flatulence)
- loss of appetite
- dry mouth
- belching
- mouth ulcer
- sweating
- muscle aches
- fever
- pain
- weight loss
- laboratory test results: changes in blood test results (such as blood chemistry and blood count)
- kidney stones
Tell your doctor as soon as possible if you notice any of the following:
- dehydration (thirst)
- diabetes (high sugar levels in the blood which may be accompanied by symptoms like frequent urination, excessive thirst, lack of energy)
- jaundice (yellowing of the skin or whites of eyes)
- muscle pain, tenderness or weakness
- hepatitis (inflammation of the liver)
The above list includes serious side effects that may require medical attention. Serious side effects are rare.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- severe or life threatening skin reaction including blisters (Stevens Johnson syndrome and Toxic Epidermal Necrolysis)
- serious allergic reaction (anaphylaxis)
- high levels of sugar in the blood
The above list includes very serious side effects. You may need urgent medical attention. These side effects are rare.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may occur in some people.
After using Norvir
Storage
Keep your tablets in the bottle until it is time to take them. If you take the tablets out of the bottle they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom, near a sink, or on a windowsill. Do not leave it in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor or pharmacist tells you to stop taking this medicine, or the medicine has passed its expiry date, ask your pharmacist what to do with any that is left over.
Product description
What it looks like
Norvir film coated tablets are white debossed with Abbott logo and the code "NK", and are supplied in bottles containing 30 tablets.
Ingredients
Norvir film coated tablets contain 100mg ritonavir with the following inactive ingredients:
- copovidone
- calcium hydrogen phosphate
- sorbitan monolaurate
- colloidal anhydrous silica
- sodium stearylfumarate
- hypromellose
- titanium dioxide
- macrogol 400
- hydroxypropylcellulose
- purified talc
- macrogol 3350
- polysorbate 80
Supplier
Norvir is supplied in Australia by:
AbbVie Pty Ltd
241 O'Riordan Street
Mascot NSW 2020
Australia
ABN: 48 156 384 262
Norvir is supplied in New Zealand by:
AbbVie Limited
6th Floor, 156-158 Victoria St
Wellington 6011
New Zealand
® Registered Trademark
Australian Registration Number
Norvir film coated tablets 100mg: AUST R 158301
This leaflet was prepared in July 2020
Version 22
Published by MIMS August 2020



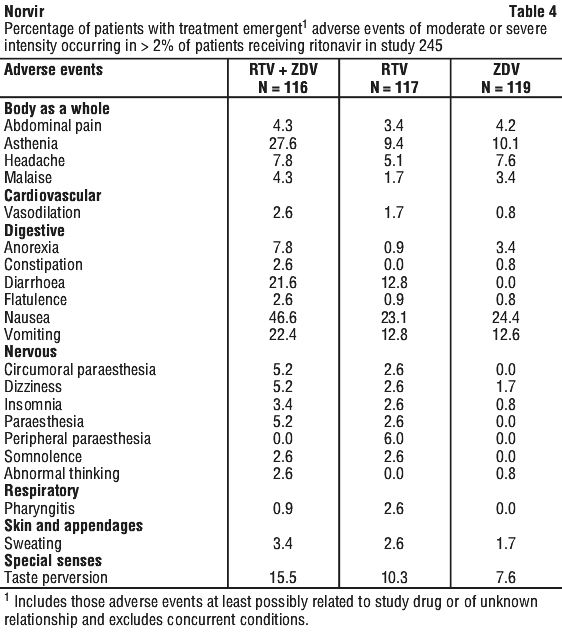
 Adverse events occurring in less than 2% of patients receiving ritonavir in all phase II/phase III studies and considered at least possibly related or unknown relationship to treatment and of at least moderate intensity are listed below by body system.
Adverse events occurring in less than 2% of patients receiving ritonavir in all phase II/phase III studies and considered at least possibly related or unknown relationship to treatment and of at least moderate intensity are listed below by body system.

 In addition, analysis of mean CD4 cell count changes from baseline over the first 16 weeks of study for the first 211 patients enrolled (mean baseline CD4 cell count = 29 cells/microlitre) showed that ritonavir was associated with larger increases in CD4 cell counts than was placebo (see Figure 2).
In addition, analysis of mean CD4 cell count changes from baseline over the first 16 weeks of study for the first 211 patients enrolled (mean baseline CD4 cell count = 29 cells/microlitre) showed that ritonavir was associated with larger increases in CD4 cell counts than was placebo (see Figure 2). Figure 3 summarises the mean changes from baseline in log HIV RNA levels for Study 247.
Figure 3 summarises the mean changes from baseline in log HIV RNA levels for Study 247.
 Figure 5 summarises the mean changes from baseline in log HIV RNA levels for Study 245.
Figure 5 summarises the mean changes from baseline in log HIV RNA levels for Study 245.


