WHAT IS IN THIS LEAFLET
This leaflet answers some common questions about Nubeqa. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using Nubeqa against the benefits your doctor expects it will have for you.
If you have any concerns about taking this medicine, ask your doctor or health care professional.
Keep this leaflet. You may need to read it again.
WHAT NUBEQA IS USED FOR
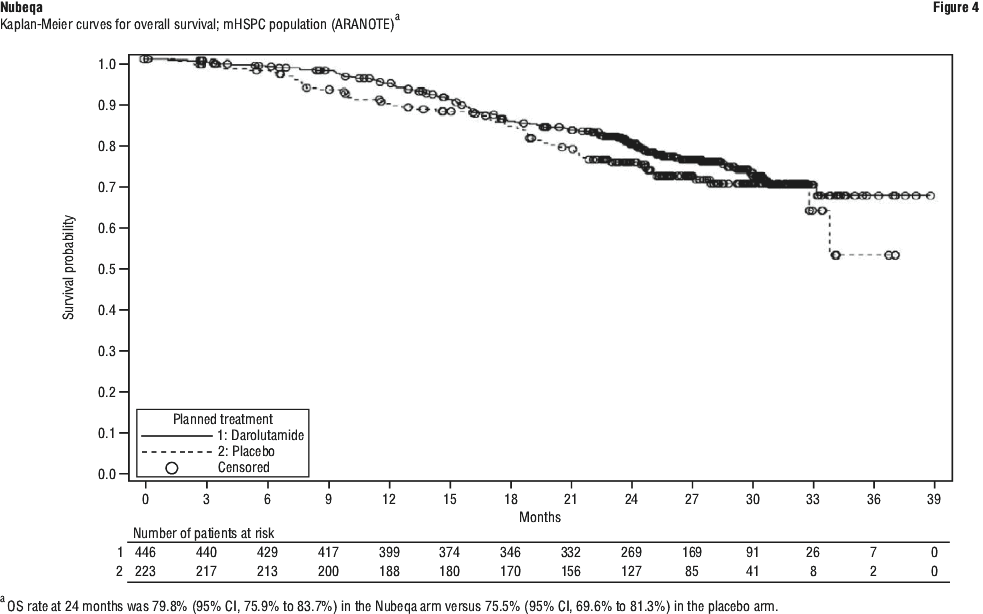
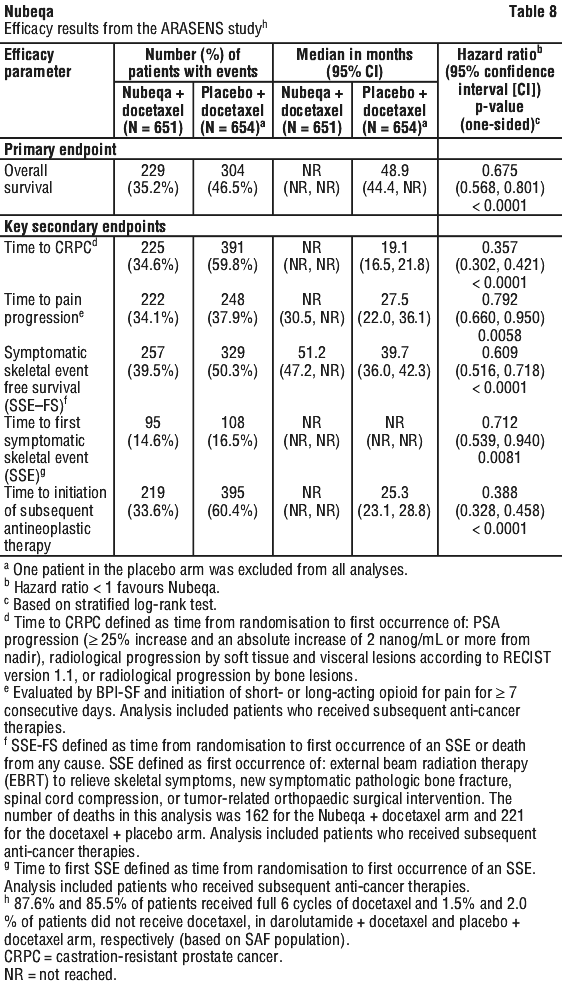
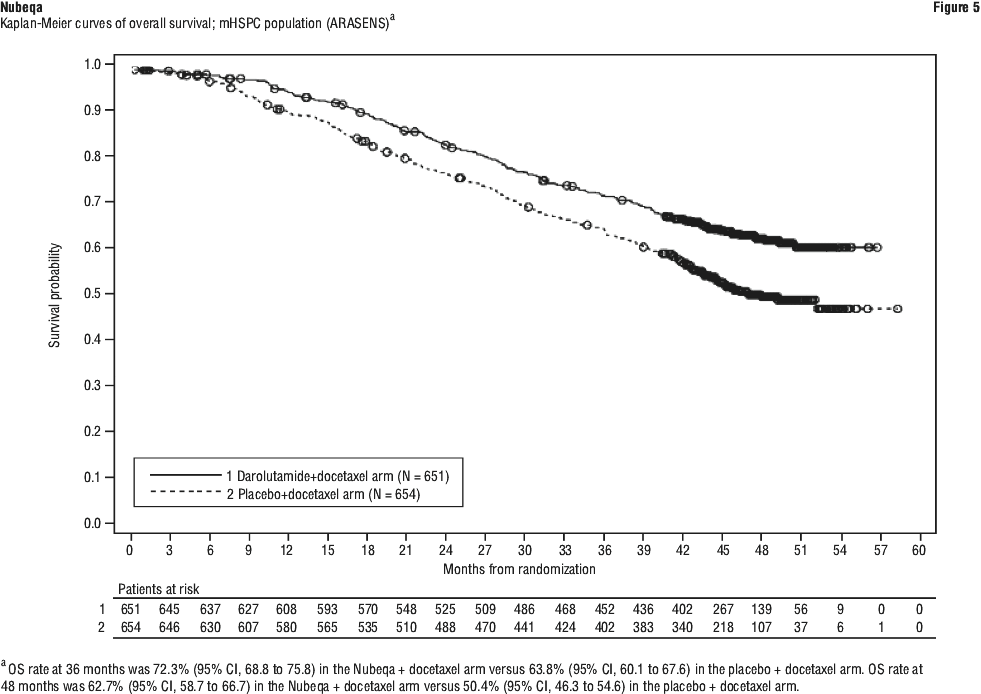
Nubeqa contains the active substance darolutamide. It is used to treat prostate cancer that has not spread to other parts of the body. Nubeqa belongs to a group of medicines called androgen receptor inhibitors. It works by blocking the activity of male sex hormones called androgens, such as testosterone leading to inhibition of growth and division of prostate cancer cells. By blocking these hormones, darolutamide stops prostate cancer cells from growing and dividing.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
BEFORE YOU TAKE NUBEQA
When you must not take Nubeqa
If you are not sure whether you should take this medicine, talk to your doctor or health care professional.
You must not take Nubeqa if:
- you have an allergy to darolutamide (the active ingredient) or any of the ingredients listed at the end of this leaflet
- the packaging is torn or shows signs of tampering
- the expiry date printed on the pack after “EXP” has passed
- You are intending to father a child. Use a highly effective method of contraception during and for 3 months after treatment with Nubeqa to prevent pregnancy.
Nubeqa is not for use in women. Do not take Nubeqa if you are pregnant, are breast-feeding or may be potentially pregnant.
If you are having sex with a pregnant woman you are advised to use a condom during and for 3 months after treatment with Nubeqa to protect the unborn baby.
The safety and efficacy in children and adolescents under 18 years of age have not been studied.
Before you start to take it
This medicine could possibly have an effect on male fertility.
Tell your doctor or health care professional. If you are intending to have children and you have questions about birth control before taking Nubeqa.
If you are not sure whether you should start using this medicine, talk to your Doctor or health care professional.
If you have not told your doctor about any of the above, tell your doctor or health care professional before you take Nubeqa.
Taking other medicines
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines. This includes medicines obtained without a prescription or even over-the-counter medicines, such as vitamins, dietary supplements or herbal medicines.
The following medicines may influence the effect of Nubeqa, or Nubeqa may influence the effect of these medicines:
- rifampicin – typically used to treat bacterial infections
- carbamazepine, phenobarbital – typically used to treat epilepsy
- St. John's Wort – typically used to treat symptoms of slightly low mood and mild anxiety
- rosuvastatin, fluvastatin, atorvastatin – typically used to treat high cholesterol
- methotrexate – typically used to treat severe joint inflammation, severe cases of the skin disease psoriasis, and cancers
- sulfasalazine – typically used to treat inflammatory bowel disease
The dose of other medicines that you are taking may need to be changed.
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket, naturopath or health food shop
HOW TO TAKE NUBEQA
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
How much to take
The recommended dose is two tablets two times a day. This is also the maximum daily dose.
How to take it
Swallow the tablets whole, take them with food.
Your doctor may also prescribe other medicines while you are taking Nubeqa.
Dose reduction
Your doctor may need to reduce your dose to one tablet two times daily or may decide to interrupt your treatment if necessary.
No dose adjustment is necessary if you have mildly impaired liver function.
No dosage adjustment is necessary if you have mildly or moderately impaired kidney function.
If you forget to take Nubeqa
Take your missed dose as soon as you remember prior to the next scheduled dose. Do not take a double dose to make up for a forgotten tablet.
If you take too much (overdose)
Continue the treatment with the next dose as scheduled.
If you think you or anybody else has taken too much Nubeqa, contact your doctor, pharmacist or the Poisons Information Centre who will advise you what to do.
You can contact the Poisons Information Centre by dialing:
- Australia: 13 11 26
- New Zealand: 0800 POISON or 0800 764 766.
If you stop taking Nubeqa
Do not stop taking this medicine unless your doctor tells you to.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
WHILE YOU ARE TAKING NUBEQA
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Nubeqa.
If you are having sex with a woman of childbearing age you are advised to use a highly effective method of contraception during and for 3 months after treatment with Nubeqa to prevent pregnancy.
If you are having sex with a pregnant woman you are advised to use a condom during and for 3 months after treatment with Nubeqa to protect the unborn baby.
Things you must not do
Do not take Nubeqa to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
SIDE EFFECTS
Tell your doctor as soon as possible if you do not feel well while you are taking Nubeqa and ask any questions you may have.
All medicines can have side effects Nubeqa helps most people, but it may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Tell your doctor if you notice any of the following and they worry you:
- fatigue (tiredness)
- rash
- pain in extremity (pain in arms and legs)
Common side effects that may show up in blood tests:
- neutrophil count decreased (reduced number of a white blood cell type called neutrophils)
- bilirubin increased (high blood levels of bilirubin, a substance produced by the liver)
- aspartate transaminase increased (high blood levels of aspartate transaminase, a substance produced by the liver)
Tell your doctor if you notice anything else that is making you feel unwell.
NUBEQA will not affect the ability to drive or use machines.
Other side effects not listed above may also occur in some people.
AFTER TAKING NUBEQA
Storage
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the carton, on each blister pack and on the bottle label after EXP. The expiry date refers to the last day of that month.
Bottle: Once the bottle is opened the medicine is stable for 3 months. Keep the bottle tightly closed after first opening.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Return any unused medicine to your pharmacist.
PRODUCT DESCRIPTION
What it looks like
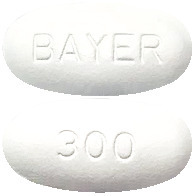
The film-coated tablets are white to off-white, oval with a length of 16 mm and a width of 8 mm. They are marked with “300” on one side, and “BAYER” on the other side.
Ingredients
Active ingredient:
- darolutamide
Inactive ingredients:
- calcium hydrogen phosphate dihydrate
- croscarmellose sodium
- hypromellose
- lactose monohydrate
- macrogol 3350
- magnesium stearate
- povidone
- titanium dioxide
Supplier
Bayer Australia Ltd
ABN 22 000 138 714
875 Pacific Highway
Pymble NSW 2073
Australian registration number
Blister AUST R 316417
Bottle AUST R 317242
Date of preparation
16 July 2020
See TGA website (www.ebs.tga.gov.au) for latest Australian Consumer Medicine Information.
® Registered Trademark of the Bayer Group, Germany.
© Bayer Australia Ltd.
All rights reserved.

Published by MIMS October 2020










 Chemical name: N-{(2S)-1-[3-(3-chloro-4-cyanophenyl)- 1H-pyrazol-1-yl]propan-2yl}- 5-(1-hydroxyethyl)-1H-pyrazole- 3-carboxamide.
Chemical name: N-{(2S)-1-[3-(3-chloro-4-cyanophenyl)- 1H-pyrazol-1-yl]propan-2yl}- 5-(1-hydroxyethyl)-1H-pyrazole- 3-carboxamide.