What is in this leaflet
Please read this leaflet carefully before you start using NUCALA.
This leaflet answers some common questions about NUCALA.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using NUCALA against the benefits he or she expects it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What NUCALA is used for
NUCALA is a medicine which contains the active substance mepolizumab, a monoclonal antibody. This antibody blocks a specific protein called interleukin-5. By blocking the action of interleukin-5, NUCALA limits the production of more eosinophils (a type of white blood cell) from the bone marrow and lowers the number of eosinophils in the bloodstream and the lungs.
NUCALA is used to treat severe eosinophilic asthma, chronic rhinosinusitis with nasal polyps and eosinophilic granulomatosis with polyangiitis (EGPA).
Severe eosinophilic asthma
Some people with severe asthma have too many eosinophils (a type of white blood cell) in the blood, lungs and tissues. Having too many eosinophils in your blood can damage the airways and can cause your asthma to get worse or can increase the number of your asthma flare ups.
NUCALA is used to treat asthma by reducing the frequency of asthma flare ups in adolescents (over 12 years of age) and adults who are already receiving asthma medicines, but whose asthma flare ups are not well controlled by medicines such as high-dose corticosteroid inhalers or beta-agonist inhalers.
NUCALA can also be used to help reduce the daily dose of oral corticosteroids in patients taking these medicines to control asthma symptoms and flare ups.
NUCALA does not treat acute asthma symptoms, such as a sudden asthma attack. Therefore, NUCALA should not be used to treat such symptoms.
Chronic rhinosinusitis with nasal polyps
Chronic rhinosinusitis with nasal polyps is a condition in which people have too many eosinophils in the blood, nose and sinuses. This can cause symptoms such as a blocked nose and loss of smell, and soft jellylike growths (called nasal polyps) to form inside the nose.
NUCALA reduces the number of eosinophils in the blood and in adults (18 years and above) can reduce the size of your polyps, relieve your nasal congestion and helps prevent surgery for nasal polyps.
NUCALA can also help reduce the need for oral corticosteroids to control your symptoms.
Eosinophilic Granulomatosis with Polyangiitis (EGPA)
EGPA is a condition where people have too many eosinophils in the blood and tissues, and also have inflammation of the blood vessels (vasculitis). EGPA most commonly affects the lungs and sinuses but often affects other organs including the skin, heart, kidneys, nerves or bowels.
NUCALA can reduce symptoms and delay a flare-up of these symptoms in people who are already taking corticosteroids.
NUCALA can also help reduce the daily dose of corticosteroids you need to control your symptoms.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you use NUCALA
Your doctor has weighed any risks of you using NUCALA against the benefits he or she expects it will have for you. You can talk to your doctor about the risks and benefits of using this medicine.
When you must not use it
Do not use NUCALA if:
- you are allergic to mepolizumab or any of the other ingredients of this medicine (listed at the end of this leaflet).
Allergic or allergic-like events often occur within minutes to hours after the medicine is administered, but in some instances symptoms can have a delayed onset of up to several days.
Some of the symptoms of an allergic reaction may include:
- chest tightness, cough, wheezing or difficulty breathing
- drop in blood pressure (fainting, dizziness, feeling lightheaded)
- swelling of the face, lips, tongue or other parts of the body
- rash, itching, hives or redness on the skin
- stomach pain or discomfort
- vomiting.
If you think any of these apply to you, do not use NUCALA until you have checked with your doctor.
If you are pregnant or if you think you may be pregnant do not use NUCALA without asking your doctor. Your doctor will consider the benefit to you and the risk to you or your baby of using NUCALA while you are pregnant.
If you plan to become pregnant, tell your doctor before starting treatment with NUCALA. Your doctor will discuss with you the benefits and potential risks of being given this medicine during pregnancy.
If you are breast-feeding, check with your doctor before you take NUCALA. It is not known whether the ingredients of NUCALA can pass into breast milk. Your doctor can discuss with you the risks and benefits involved.
For severe eosinophilic asthma, NUCALA is not recommended for children aged under 12 years or in adolescents who weigh less than 45 kg, as the safety and effectiveness are not known in this population.
For Nasal Polyps and EGPA, NUCALA may be used in adults(18 years and above) only. NUCALA has not been tested in children with Nasal Polyps or EGPA.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start using this medicine, talk to your doctor.
Before you start to use it
Your doctor should give you a Personal Action Plan to help manage your asthma. This plan will include what medicines to take regularly to control your asthma, as well as what "reliever" medicines to use when you have sudden attacks of breathlessness or wheezing.
Ask your doctor or pharmacist if you have any questions about your Action Plan.
Talk to your doctor before you use NUCALA if:
- you have had an allergic reaction before
- you have an existing infection or live in a region where infections caused by parasites are common or if you are travelling to such a region as NUCALA may weaken your resistance to such infections. Parasitic infections should be treated prior to starting treatment with NUCALA.
You may need extra check-ups while you are being treated with NUCALA.
NUCALA does not treat acute asthma symptoms, such as a sudden asthma flare up. Therefore, NUCALA should not be used to treat such symptoms.
Asthma-related side effects or flare ups may occur during treatment with NUCALA.
If your asthma symptoms get worse while receiving injections of NUCALA speak to your doctor.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop. This should include all of the medicines that you are using for your asthma.
Do not suddenly stop taking your corticosteroids once you have started NUCALA. Corticosteroids must be stopped gradually, under the supervision of your doctor.
How to use NUCALA
NUCALA is given to you by a healthcare professional, doctor, nurse or pharmacist as an injection just under the skin (subcutaneously).
How much to use
Severe eosinophilic asthma:
The recommended dose is 100 mg. You will be given 1 injection, once every four weeks.
Nasal Polyps:
Adults aged 18 years and over:
The recommended dose is 100 mg. You will be given 1 injection, once every four weeks.
EGPA:
Adults aged 18 years and over:
The recommended dose is 300 mg. You will be given 3 injections once every four weeks.
If you forget to take it
If a dose of NUCALA is missed contact your doctor or hospital as soon as possible to re-schedule your appointment.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
In Australia, immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, if you think that you or anyone else may have taken too much NUCALA. Do this even if there are no signs of discomfort or poisoning.
While you are using NUCALA
Things you must do
If you have an Action Plan for your asthma that you have agreed with your doctor, follow it closely at all times.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are using NUCALA.
Tell any other doctors, dentists, and pharmacists who treat you that you are using this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are using this medicine.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not stop receiving injections of NUCALA unless your doctor tells you to. Interrupting or stopping the treatment with NUCALA may cause your asthma symptoms and flare ups to come back or occur more frequently.
If you have any further questions on the use of this medicine, ask your doctor, pharmacist or nurse.
Things to be careful of
This medicine is not expected to affect your ability to drive a car or operate machinery. However, it is prudent to be careful with driving or operating machinery until you know how NUCALA affects you.
Side effects
Like all medicines, NUCALA can cause side effects, although not everybody gets them. The side effects caused by NUCALA are usually mild to moderate but can occasionally be serious.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Very common side effects
This may affect more than 1 in 10 people:
- headache
Common side effects
These may affect up to 1 in 10 people:
- injection-site reaction (pain, redness, swelling, itching, and burning sensation of the skin near where the injection was given)
- eczema (itchy red patches on the skin)
- back pain
- fatigue (tiredness)
- pharyngitis (sore throat)
- lower respiratory tract infection (congestion, cough, discomfort)
- nasal congestion (stuffy nose)
- upper abdominal pain (stomach pain or discomfort in the upper area of the stomach)
- urinary tract infection (blood in urine, painful and frequent urination, fever, pain in lower back)
- fever (high temperature).
- Some side effects may occur more frequently in people with EGPA, including headache, injection site reactions, diarrhoea and vomiting.
Tell your doctor or pharmacist if you experience any of the side effects listed, particularly if they become severe or troublesome, or if you notice any side effects not listed in this leaflet.
If you think you are having an allergic reaction to NUCALA, stop using this medicine and tell your doctor or a nurse immediately or go to the Emergency Department at your nearest hospital.
Symptoms of an allergic reaction usually include some or all of the following:
- chest tightness, cough, wheezing or difficulty breathing
- drop in blood pressure (fainting, dizziness, feeling lightheaded)
- swelling of the face, lips, tongue or other parts of the body
- rash, itching, hives, or redness on the skin
- stomach pain or discomfort
- vomiting
If you get any side effects, talk to your doctor, pharmacist or nurse.
Other side effects not listed above may occur in some people.
Storage
Do not use NUCALA after the expiry date shown on the pack. The expiry date refers to the last day of that month.
Unopened vials:
Refer to the product carton, which will state either:
"Store at 2°C to 8°C (Refrigerate. Do not freeze)".
OR
"Store below 25°C (Do not freeze)".
Keep the vial in the outer carton in order to protect from light.
Reconstituted solution:
Store below 25°C.
Reconstituted solution does not need to be protected from light. It is stable for up to 6 hours. Discard after 6 hours if not used.
Do not store NUCALA or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car.
Keep it where children cannot reach it.
Disposal
If your doctor tells you to stop using this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
NUCALA 100 mg powder for injection is a sterile, white powder supplied in single-use, clear, colourless type I glass vial with a rubber stopper.
NUCALA 100 mg powder for injection is available in a pack containing 1 single-use vial.
Ingredients
The active ingredient in NUCALA is mepolizumab.
Each dose contains 100 mg of the active ingredient mepolizumab.
NUCALA also contains the inactive ingredients:
- sucrose
- dibasic sodium phosphate heptahydrate
- polysorbate 80
Supplier
NUCALA is supplied in Australia by:
GlaxoSmithKline Australia Pty Ltd
Level 4, 436 Johnston Street
Abbotsford Victoria 3067
Australia.
Where to go for further information
Pharmaceutical companies are not in a position to give people an individual diagnosis or medical advice. Your doctor or pharmacist is the best person to give you advice on the treatment of your condition. You may also be able to find general information about your disease and its treatment from patient information groups.
This leaflet was prepared on 10 January 2022.
The information provided applies only to: NUCALA powder for injection.
Powder for injection: AUST R 232028
Trade marks are owned by or licensed to the GSK group of companies.
© 2022 GSK group of companies or its licensor.
Version 6.0
Published by MIMS June 2025






 Additionally, health related quality of life was measured using SGRQ. At Week 24, there was a statistically significant improvement in the mean SGRQ score for mepolizumab compared with placebo: -5.8 (95% CI: -10.6, -1.0; p = 0.019). At Week 24, the proportion of subjects with a clinically meaningful decrease in SGRQ score (defined as a decrease of at least 4 units from baseline) was greater for mepolizumab (58%, 40/69) compared with placebo (41%, 27/66).
Additionally, health related quality of life was measured using SGRQ. At Week 24, there was a statistically significant improvement in the mean SGRQ score for mepolizumab compared with placebo: -5.8 (95% CI: -10.6, -1.0; p = 0.019). At Week 24, the proportion of subjects with a clinically meaningful decrease in SGRQ score (defined as a decrease of at least 4 units from baseline) was greater for mepolizumab (58%, 40/69) compared with placebo (41%, 27/66). The co-primary endpoints were change from baseline in total endoscopic NP score at Week 52 and change from baseline in mean nasal obstruction VAS score during weeks 49-52.
The co-primary endpoints were change from baseline in total endoscopic NP score at Week 52 and change from baseline in mean nasal obstruction VAS score during weeks 49-52. All secondary endpoints were statistically significant and provided support for the co-primary endpoints. The key secondary endpoint was the time to first NP surgery up to Week 52 (see Figure 1). Data from the other secondary endpoints are presented in Table 10.
All secondary endpoints were statistically significant and provided support for the co-primary endpoints. The key secondary endpoint was the time to first NP surgery up to Week 52 (see Figure 1). Data from the other secondary endpoints are presented in Table 10.


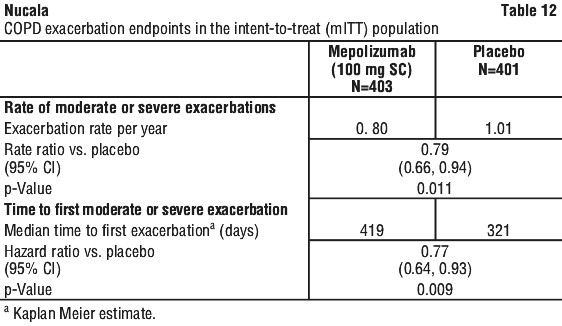
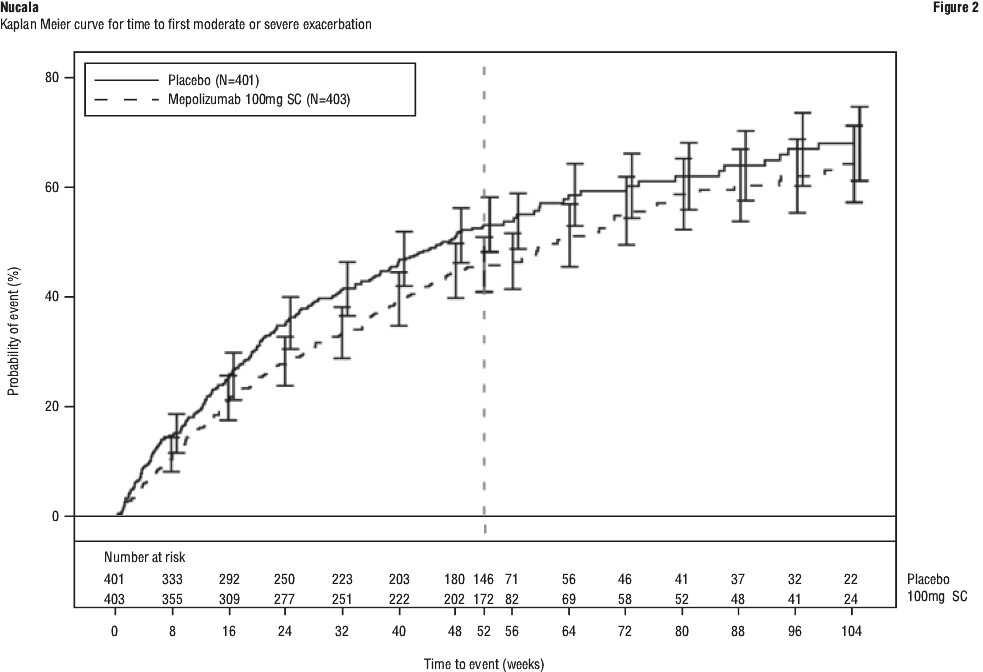
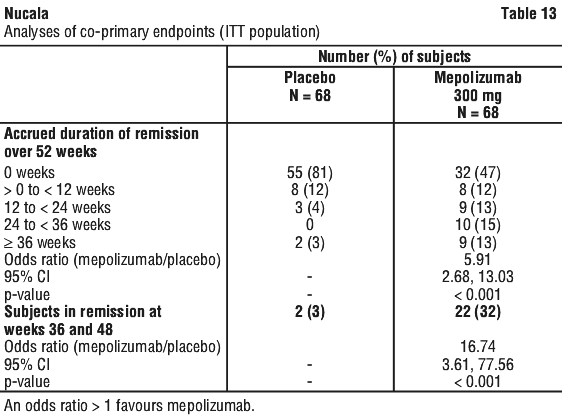 Additionally, a statistically significant benefit for these endpoints was demonstrated using remission defined as BVAS = 0 plus prednisolone/prednisone ≤ 7.5 mg/day. There was a greater accrued time in remission in the mepolizumab group compared with placebo, in subjects with a baseline blood eosinophil count (BEC) ≥ 150 cells/microL.
Additionally, a statistically significant benefit for these endpoints was demonstrated using remission defined as BVAS = 0 plus prednisolone/prednisone ≤ 7.5 mg/day. There was a greater accrued time in remission in the mepolizumab group compared with placebo, in subjects with a baseline blood eosinophil count (BEC) ≥ 150 cells/microL. The total estimated molecular weight for mepolizumab is 149 kDa.
The total estimated molecular weight for mepolizumab is 149 kDa.