1 Name of Medicine
Rimegepant.
2 Qualitative and Quantitative Composition
Each orally disintegrating tablet (ODT) contains rimegepant sulfate, equivalent to 75 mg rimegepant.
Excipients with known effect.
Sucralose, mannitol.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Orally disintegrating tablet.
White to off-white, circular, diameter 14 mm and debossed with a symbol consisting of two quadrants.
4.1 Therapeutic Indications
Nurtec ODT is indicated for:
acute treatment of migraine with or without aura in adults;
prophylactic treatment of episodic migraine in adults who have at least 4 migraine attacks per month.
4.2 Dose and Method of Administration
Dosage.
Acute treatment of migraine.
The recommended dose is 75 mg rimegepant orally disintegrating tablet, as needed. The maximum dose in a 24-hour period is 75 mg.
Prophylaxis of migraine.
The recommended dose is 75 mg rimegepant every other day. If also requiring rimegepant for the acute treatment of migraine, do not exceed a total dose of 75 mg rimegepant in a 24-hour period.
Nurtec ODT can be taken with or without meals.
Method of administration.
Nurtec ODT is for oral use.
The orally disintegrating tablet should be placed on the tongue or under the tongue. It will disintegrate in the mouth and can be taken without liquid.
Patients should be advised to use dry hands when opening the blister and refer to the Consumer Medicines Information for complete instructions.
Dosage adjustment.
Elderly (aged 65 and over).
There is limited experience with rimegepant in patients aged 65 years or older. No dose adjustment is required as the pharmacokinetics of rimegepant are not affected by age (see Section 5.2 Pharmacokinetic Properties).
Renal impairment.
No dose adjustment is required in patients with mild, moderate, or severe renal impairment. Severe renal impairment resulted in a > 2-fold increase in unbound AUC but less than a 50% increase in total AUC (see Section 5.2 Pharmacokinetic Properties). Rimegepant has not been studied in patients with end-stage renal disease and in patients on dialysis. Use of rimegepant in patients with end-stage renal disease (CLcr < 15 mL/min) should be avoided.
Hepatic impairment.
No dose adjustment is required in patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment. Plasma concentrations (unbound AUC) of rimegepant were significantly higher in subjects with severe (Child-Pugh C) hepatic impairment (see Section 5.2 Pharmacokinetic Properties). The use of rimegepant in patients with severe hepatic impairment should be avoided.
Paediatric population.
The safety and efficacy of Nurtec ODT in paediatric patients (< 18 years of age) have not been established. No data are available.
Concomitant medicinal products.
Another dose of rimegepant should be avoided within 48 hours when it is concomitantly administered with moderate inhibitors of CYP3A4 or with strong inhibitors of P-gp.
Avoid concomitant administration of Nurtec ODT with strong inhibitors of CYP3A4, strong or moderate inducers of CYP3A4.
See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.4.3 Contraindications
Hypersensitivity to rimegepant or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Hypersensitivity reactions, including dyspnoea and rash, have occurred in less than 1% of patients treated with rimegepant in clinical studies. Hypersensitivity reactions, including serious hypersensitivity such as anaphylactic reaction, have been reported in the clinical and post-marketing settings (see Section 4.8 Adverse Effects Undesirable Effects)). Some hypersensitivity reactions can occur days after administration. If a hypersensitivity reaction occurs, rimegepant should be discontinued and appropriate therapy should be initiated.
There are limited data supporting the efficacy and safety of:
Additional daily doses of rimegepant for acute treatment of the same migraine attack.
Rimegepant for the acute treatment of migraine attacks occurring whilst taking a CGRP blocking monoclonal antibody as prophylaxis.
There are limited data supporting the efficacy of rimegepant for the acute treatment of migraine attacks occurring whilst using rimegepant as prophylaxis.
The benefit and the safety of using ≥ 18 doses of rimegepant per month is not established.
Rimegepant clinical trials generally excluded participants with new or unstable cardiovascular disease, uncontrolled hypertension and uncontrolled diabetes (see Section 5.1 Pharmacodynamic Properties).
Medication overuse headache (MOH).
Overuse of many medicinal products for headaches can make them worse. Although there is no evidence that use of rimegepant up to once daily can lead to MOH, the diagnosis of MOH should be suspected in patients who have frequent or daily headaches despite (or because of) the regular use of medicinal products for acute headache. If this situation is experienced or suspected, medical advice should be obtained, and treatment should be discontinued.
Use in hepatic impairment.
Nurtec ODT is not recommended in patients with severe hepatic impairment. See Section 4.2 Dose and Method of Administration.
Use in renal impairment.
Nurtec ODT is not recommended in patients with end-stage renal disease (CLcr < 15 mL/min). See Section 4.2 Dose and Method of Administration.
Use with strong inhibitors of CYP3A4.
Nurtec ODT is not recommended for concomitant use with strong inhibitors of CYP3A4. See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
Use with moderate and strong inducers of CYP3A4.
Nurtec ODT is not recommended for concomitant use with strong or moderate inducers of CYP3A4. See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
Use in the elderly.
There is limited experience with rimegepant in patients aged 65 years or older. No dose adjustment is required as the pharmacokinetics of rimegepant are not affected by age (see Section 5.2 Pharmacokinetic Properties).
Paediatric use.
The safety and efficacy of Nurtec ODT in paediatric patients (< 18 years of age) have not been established. No data are available.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
In vitro assessment of drug interactions.
Based on in vitro studies, rimegepant is a substrate of CYP3A4 and CY2C9. Rimegepant is not an inhibitor of CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or UGT1A1 at clinically relevant concentrations. However, rimegepant is a weak inhibitor of CYP3A4 with time-dependent inhibition. Rimegepant is not an inducer of CYP1A2, CYP2B6, or CYP3A4 at clinically relevant concentrations.
In vitro, rimegepant is a substrate of P-gp and BCRP efflux transporters. Rimegepant is not a substrate of OATP1B1 or OATP1B3. Considering its low renal clearance, rimegepant was not evaluated as a substrate of the OAT1, OAT3, OCT2, MATE1, or MATE2-K.
Rimegepant is not an inhibitor of P-gp, systemic BCRP, OAT1, or MATE2-K at clinically relevant concentrations. It is a weak inhibitor of OATP1B1, intestinal BCRP and OAT3. Rimegepant is an inhibitor of OATP1B3, OCT2, and MATE1.
In vivo studies.
Enzymes. Effect of CYP3A4 inhibitors on rimegepant.
Inhibitors of CYP3A4 increase plasma concentrations of rimegepant. Concomitant administration of rimegepant with strong CYP3A4 inhibitors (e.g. clarithromycin, itraconazole, ritonavir) is not recommended (see Section 4.4 Special Warnings and Precautions for Use). Concomitant administration of rimegepant with itraconazole resulted in a significant increase in rimegepant exposure (AUC by 4-fold and Cmax by 1.5-fold).
Concomitant administration of rimegepant with medicinal products that moderately inhibit CYP3A4 (e.g. diltiazem, erythromycin, fluconazole) may increase exposure to rimegepant. Concomitant administration of rimegepant with fluconazole resulted in increased exposures of rimegepant (AUC by 1.8-fold) with no relevant effect on Cmax. Another dose of rimegepant within 48 hours should be avoided when it is concomitantly administered with moderate inhibitors of CYP3A4 (e.g. fluconazole) (see Section 4.2 Dose and Method of Administration).
Effect of CYP3A4 inducers on rimegepant.
Inducers of CYP3A4 decrease plasma concentrations of rimegepant. Concomitant administration of Nurtec ODT with strong CYP3A4 inducers (e.g. phenobarbital, rifampicin, St John's wort (Hypericum perforatum)) or moderate CYP3A4 inducers (e.g. bosentan, efavirenz, modafinil) is not recommended (see Section 4.4 Special Warnings and Precautions for Use). The effect of CYP3A4 induction may last for up to 2 weeks after discontinuation of the strong or moderate CYP3A4 inducer. Concomitant administration of rimegepant with rifampicin resulted in a significant decrease (AUC reduced by 80% and Cmax by 64%) in rimegepant exposure, which may lead to loss of efficacy.
Transporters. Effect of P-gp and BCRP inhibitors on rimegepant.
Inhibitors of P-gp and BCRP efflux transporters may increase plasma concentrations of rimegepant. Another dose of Nurtec ODT within 48 hours should be avoided when it is concomitantly administered with strong inhibitors of P-gp (e.g. ciclosporin, verapamil, quinidine). Concomitant administration of rimegepant with ciclosporin (a potent P-gp and BCRP inhibitor) or with quinidine (a selective P-gp inhibitor) resulted in a significant increase of similar magnitude in rimegepant exposure (AUC and Cmax by > 50%, but less than two-fold). Therefore, concomitant administration of rimegepant with BCRP inhibitors is not expected to have a clinically significant impact on rimegepant exposures.
Concomitant administration of rimegepant with metformin, a MATE1 transporter substrate, resulted in no clinically significant impact on either metformin pharmacokinetics or on glucose utilisation. No clinical drug interactions are expected for rimegepant with OATP1B3 or OCT2, at clinically relevant concentrations.
Other medicinal products.
No significant pharmacokinetic interactions were observed when Nurtec ODT was concomitantly administered with oral contraceptives (norelgestromin, ethinyl estradiol), midazolam (a sensitive CYP3A4 substrate), or sumatriptan.
No clinically relevant differences in resting blood pressure were observed when rimegepant was concomitantly administered with sumatriptan (12 mg subcutaneous, given as two 6 mg doses separated by one hour) compared with sumatriptan alone to healthy volunteers.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Animal studies showed no clinically relevant impact on female and male fertility. In an oral fertility study in rats, rimegepant-related effects were noted only at the high dose of 150 mg/kg/day (decreased fertility and increased pre-implantation loss) that produced maternal toxicity and estimated systemic exposures > 80x the human AUC at 75 mg/day. There was no impact on either male or female fertility with rimegepant doses up to 60 mg/kg/day (estimated exposures approximately 43x (males) and 71x (females) the human AUC at 75 mg/day).
(Category B1)
There are limited data from the use of rimegepant in pregnant women. Animal studies demonstrate that rimegepant does not result in embryofetal death or fetal malformations at clinically relevant exposures. There were no developmental effects in rats at oral doses up to 60 mg/kg/day (exposures 40x the human AUC at 75 mg/day) or in rabbits at up to the highest oral dose tested of 50 mg/kg/day (exposures 8x the human AUC at 75 mg/day). As a precautionary measure, it is preferable to avoid the use of Nurtec ODT during pregnancy.
A lactation study was conducted in 12 breast-feeding women who were between 2 weeks and 6 months post-partum and were administered a single dose of rimegepant 75 mg. The results have established an average milk-to-plasma ratio of 0.20 and a relative infant dose of less than 1% of the maternal weight-adjusted dose. These data support that transfer of rimegepant into breastmilk is low. There are no data on the effects of rimegepant on a breastfed infant or on milk production.
The developmental and health benefits of breast-feeding should be considered along with the mother's clinical need for Nurtec ODT and any potential adverse reactions on the breastfed infant from rimegepant or from the underlying maternal condition.4.7 Effects on Ability to Drive and Use Machines
Drowsiness following rimegepant has been infrequently reported and at a similar incidence as placebo. However, migraine may cause drowsiness in some patients. Therefore, caution is recommended in patients driving or using machines during a migraine attack, including following treatment.
4.8 Adverse Effects (Undesirable Effects)
The efficacy of Nurtec ODT for the acute treatment of migraine with and without aura in adults was studied in three randomised, double-blind, placebo-controlled trials (Studies 1-3). In these studies, 3,551 unique patients received a single dose of rimegepant 75 mg (N=1,771) or placebo (N=1,782).
The efficacy of rimegepant was evaluated as a prophylactic treatment for migraine in a randomised, double blind, placebo-controlled study (Study 4). In this study, 741 patients received rimegepant (N=370) or placebo (N=371) every other day during the 12-week double blind treatment period of the study. During double blind treatment period of the study, patients treated with rimegepant or placebo received a mean (SD) / median of 13.8 (1.55) / 14.2 and 13.9 (1.79) / 14.2 tablets per month, respectively.
In patients (N=671) from this study who entered the 52-week open-label extension period of the study and received at least 1 dose of rimegepant during the double blind treatment or open-label extension of the study, patients were on rimegepant for a mean (SD) / median of 44.6 (20.51) / 51.6 weeks. The mean (SD) / median number of tablets of rimegepant received by these patients per month was 14.3 (2.10) /14.2.
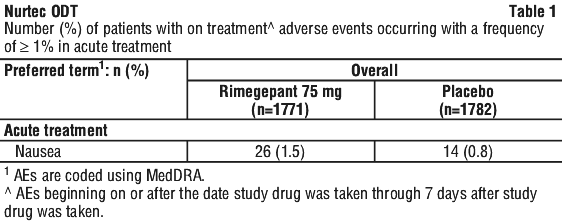
The most common adverse reaction was nausea for acute treatment (1.2%) and for migraine prophylaxis (1.4%). Most of the reactions were mild or moderate in severity.
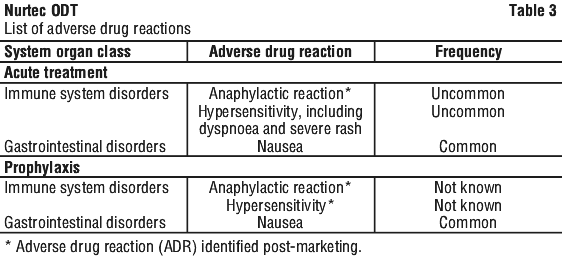
Hypersensitivity, including dyspnoea and severe rash, occurred in less than 1% of patients treated.
A tabulated summary of on treatment adverse events occurring in ≥ 1% of patients treated with rimegepant for acute treatment during double blind treatment is provided in Table 1.
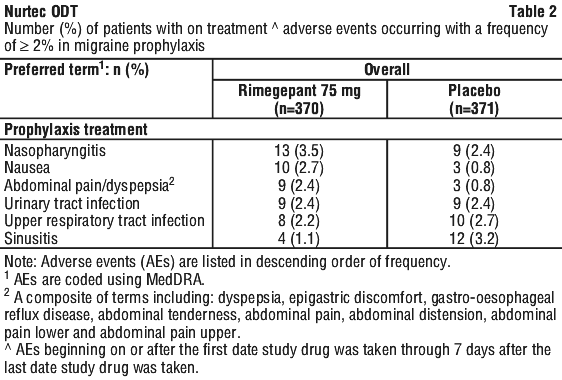
 A tabulated summary of on treatment adverse events occurring in ≥ 2% of patients treated with rimegepant for migraine prophylaxis during double blind treatment is provided in Table 2.
A tabulated summary of on treatment adverse events occurring in ≥ 2% of patients treated with rimegepant for migraine prophylaxis during double blind treatment is provided in Table 2.
 Adverse reactions are listed by MedDRA system organ class in Table 3. The corresponding frequency category for each drug reaction is based on the following: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from the available data).
Adverse reactions are listed by MedDRA system organ class in Table 3. The corresponding frequency category for each drug reaction is based on the following: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from the available data).
Long-term safety.
Long-term safety of rimegepant was assessed in two one-year, open-label extensions; 1662 patients received rimegepant for at least 6 months and 740 received rimegepant for 12 months for acute or prophylactic treatment.
Description of selected adverse reactions.
Hypersensitivity reactions.
Hypersensitivity, including dyspnoea and severe rash, occurred in less than 1% of patients treated in clinical studies. Hypersensitivity reactions can occur days after administration, and delayed serious hypersensitivity has occurred.
Post marketing experience.
The limited post-marketing experience with this formulation of rimegepant is consistent with the above profile.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/safety/reporting-problems.4.9 Overdose
There is limited clinical experience with rimegepant overdose. No overdose symptoms have been reported. Treatment of an overdose of rimegepant should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. No specific antidote for the treatment of rimegepant overdose is available. Rimegepant is unlikely to be significantly removed by dialysis because of high serum protein binding.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Rimegepant binds with high affinity to the human calcitonin gene-related peptide (CGRP) receptor and antagonises CGRP receptor function.
Multiple lines of clinical evidence point to a role for CGRP in migraine pathophysiology: 1) serum levels of CGRP are elevated during migraine; 2) treatment with anti-migraine drugs returns CGRP levels to normal coincident with pain relief and 3) intravenous (IV) CGRP infusion produces lasting pain in non-migraineurs and migraineurs.
Clinical trials.
The studies for acute and prophylactic treatment excluded patients with evidence of uncontrolled, unstable or recently diagnosed cardiovascular disease, such as ischaemic heart disease, coronary artery vasospasm, and cerebral ischaemia. Patients with myocardial infarction (MI), acute coronary syndrome (ACS), percutaneous coronary intervention (PCI), cardiac surgery, stroke or transient ischaemic attack (TIA) during the 6 months prior to screening were excluded.
In controlled clinical trials, participants on other CGRP antagonists or with uncontrolled hypertension and/or diabetes were excluded. The concomitant use of CGRP antibodies for prophylaxis was permitted in a cohort of participants in the long-term safety study comprising of 13 subjects.
Acute treatment.
The efficacy of Nurtec ODT for the acute treatment of migraine with and without aura in adults was studied in three randomised, double blind, placebo-controlled trials (Studies 1-3). Patients were instructed to treat a migraine of moderate to severe headache pain intensity. Rescue medicinal products (i.e. NSAIDs, paracetamol, and/or an antiemetic) was allowed 2 hours after the initial treatment. Other forms of rescue medicinal products such as triptans were not allowed within 48 hours of initial treatment. Approximately 14% of patients were taking preventive medicinal products for migraine at baseline. None of the patients in Study 1 were on concomitant preventive medicinal products that act on the calcitonin gene-related peptide pathway.
In the 3 pivotal trials for the acute treatment of migraine, demographic and baseline characteristics were representative of the potential treatment population. The median age ranged from 38.9 to 41.5 years across treatment groups. Most subjects were female (84.9% to 89.2% across treatment groups) and white (73.4% to 82.1% across treatment groups). About 1% of subjects in all treatment groups had a CV risk contraindicating triptan use (range 0.4% to 1.2%). Almost half of the subjects (range 41.4% to 52.7% across treatment groups) had a body mass index (BMI) ≥ 30 kg/m2. The median number of moderate or severe pain intensity migraine attacks per month was 4.0 for all treatment groups. Across treatment groups, most subjects' primary migraine type was migraine without aura (range 65.0% to 71.7%).
The primary efficacy analyses were conducted in patients who treated a migraine with moderate to severe pain. Pain freedom and MBS were the co-primary endpoints. Pain freedom was defined as a reduction of moderate or severe headache pain to no headache pain, and most bothersome symptom (MBS) freedom was defined as the absence of the self-identified MBS (i.e. photophobia, phonophobia, or nausea). Among patients who selected an MBS, the most commonly selected symptom was photophobia (54%), followed by nausea (28%), and phonophobia (15%).
In Study 1, the percentage of patients achieving headache pain freedom and MBS freedom at 2 hours after a single dose was statistically significantly greater in patients who received Nurtec ODT compared to those who received placebo (Table 4). In addition, statistically significant effects of Nurtec ODT compared to placebo were demonstrated for the additional efficacy endpoints of pain relief at 2 hours, sustained pain freedom from 2 to 48 hours, use of rescue medication within 24 hours, and ability to function normally at 2 hours after dosing. Pain relief was defined as a reduction in migraine pain from moderate or severe severity to mild or none. Pivotal single attack, double blind, placebo-controlled studies 2 and 3 were conducted in patients with migraine who received one 75 mg rimegepant bioequivalent dosage form.
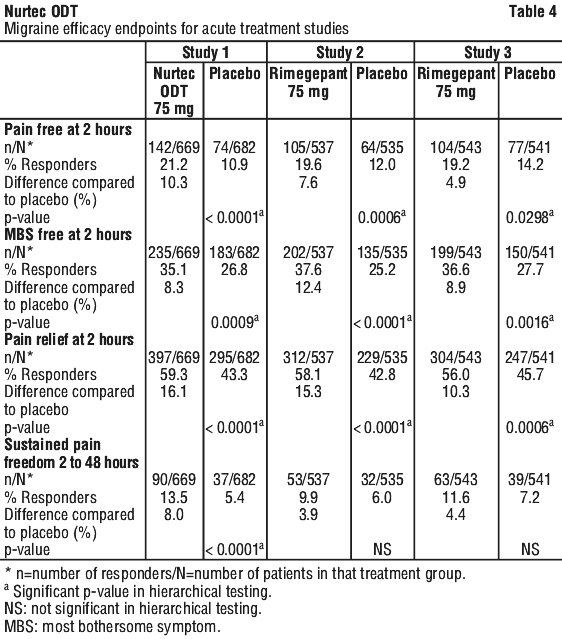 Figure 1 presents the percentage of patients achieving migraine pain freedom within 2 hours following treatment in Study 1.
Figure 1 presents the percentage of patients achieving migraine pain freedom within 2 hours following treatment in Study 1.
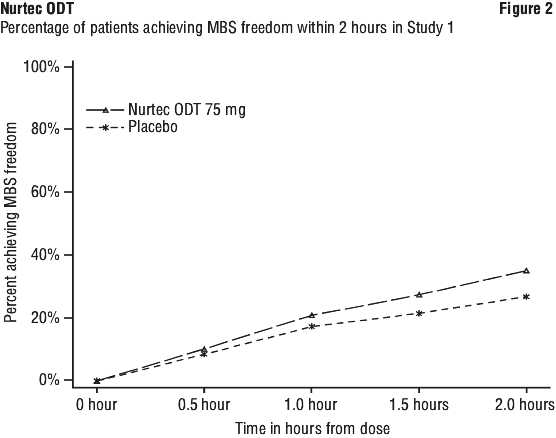
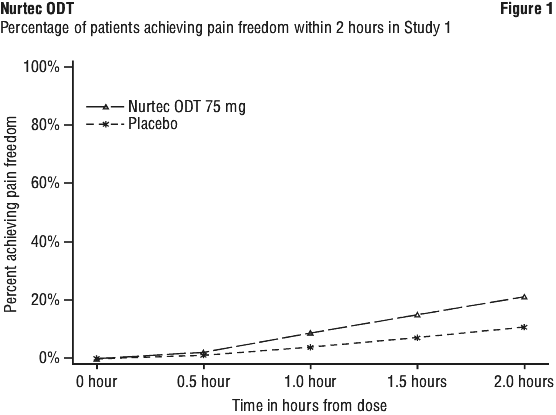 Figure 2 presents the percentage of patients achieving MBS freedom within 2 hours in Study 1.
Figure 2 presents the percentage of patients achieving MBS freedom within 2 hours in Study 1.
 The incidence of photophobia and phonophobia was reduced at 2 hours following administration of Nurtec ODT 75 mg as compared to placebo in all 3 studies.
The incidence of photophobia and phonophobia was reduced at 2 hours following administration of Nurtec ODT 75 mg as compared to placebo in all 3 studies.
Prophylaxis.
The efficacy of rimegepant was evaluated as a prophylactic treatment of migraine in a randomised, double blind, placebo-controlled study (Study 4).
Study 4 included male and female adults with at least a 1-year history of migraine (with or without aura). Patients had a history of 4 to 18 migraine attacks of moderate to severe pain intensity per 4-week period within the 12 weeks prior to the screening visit. Patients experienced an average of 10.9 headache days during the 28-day observational period, which included an average of 10.2 migraine days, prior to randomisation into the study. The study randomised patients to receive rimegepant 75 mg (N=373) or placebo (N=374) for up to 12 weeks. Patients were instructed to take randomised treatment once every other day (EOD) for the 12-week treatment period. Patients were allowed to use other acute treatments for migraine (e.g. triptans, NSAIDs, paracetamol, antiemetics) as needed. Approximately 22% of patients were taking preventive medicinal products for migraine at baseline. Patients were allowed to continue in an open-label extension study for an additional 12 months.
In the pivotal trial for migraine prophylaxis, demographic and baseline characteristics representative of the potential treatment population and were well balanced across the rimegepant and placebo groups. Among treated subjects, the median age was 40.0 years, the majority of subjects were female (82.7%), and most subjects were white (81.5%). Approximately 10% of evaluable mITT subjects were taking preventive medications for migraine at baseline. The median age at migraine disease onset was 18.0 years, the median number of moderate to severe migraine attacks per month was 8.0 (range: 4 to 18), and the median average duration of untreated migraine attacks was 24.0 hours. The primary migraine type was migraine without aura (60.1%). Of the evaluable subjects, 22.7% met the ICHD-3 criteria for chronic migraine.
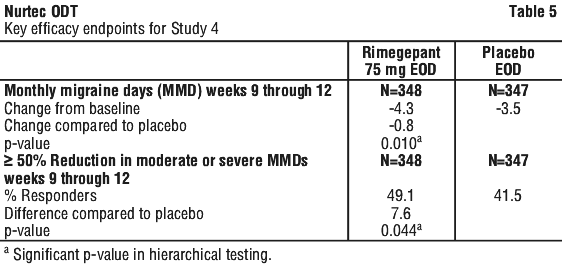
The primary efficacy endpoint for Study 4 was the change from baseline in the mean number of monthly migraine days (MMDs) during Weeks 9 through 12 of the double blind treatment phase. Secondary endpoints included the achievement of a ≥ 50% reduction from baseline in monthly moderate or severe migraine days.
Rimegepant 75 mg dosed EOD demonstrated statistically significant improvements for key efficacy endpoints compared to placebo, as summarised in Table 5 and shown graphically in Figure 3.
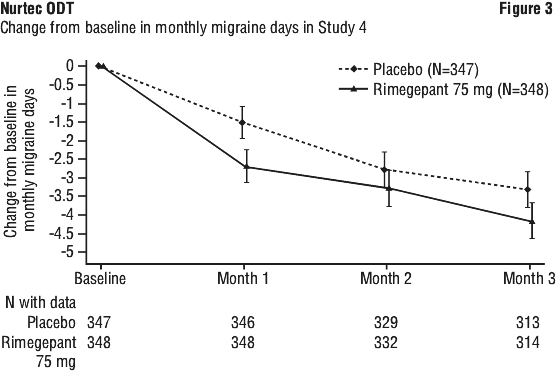
Long-term efficacy.
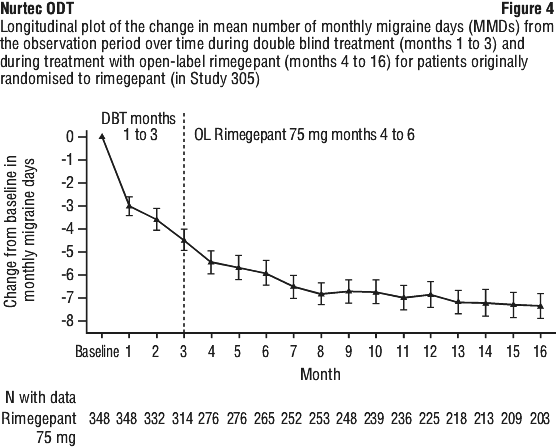
Patients participating in Study 4 were allowed to continue in an open-label extension study for an additional 12 months. Efficacy was sustained for up to 1 year in an open-label study extension in which patients received rimegepant 75 mg every other day plus as needed on nonscheduled dosing days (Figure 4). A portion composed of 203 patients assigned to rimegepant completed the overall 16-month treatment period. In these patients, the overall mean reduction from baseline in the number of MMDs averaged over the 16-month treatment period was 6.2 days. For the open label extension phase in Figure 4, the data presented as OL Rimegepant 75 mg Months 4 to 16 have no comparator group. The data are suggestive of maintenance of effect of rimegepant to 16 months.
5.2 Pharmacokinetic Properties
Absorption.
Following oral administration, rimegepant is absorbed with the maximum concentration at 1.5 hours. Following a supratherapeutic dose of 300 mg, the absolute oral bioavailability of rimegepant was approximately 64%.
Effects of food.
Following administration of rimegepant under fed conditions with a high-fat or low-fat meal, Tmax was delayed by 1 to 1.5 hours. A high-fat meal reduced Cmax by 42 to 53% and AUC by 32 to 38%. A low-fat meal reduced Cmax by 36% and AUC by 28%. Rimegepant was administered without regard to food in clinical safety and efficacy studies.
The impact of the reduction in rimegepant exposure because of administration with food on its efficacy is unknown.
Distribution.
The steady state volume of distribution of rimegepant is 120 L. Plasma protein binding of rimegepant is approximately 96%.
Metabolism.
Rimegepant is primarily metabolised by CYP3A4 and to a lesser extent by CYP2C9. Rimegepant is the primary form (~77%) with no major metabolites (i.e. > 10%) detected in plasma.
Excretion.
The elimination half-life of rimegepant is approximately 11 hours in healthy subjects. Following oral administration of [14C]-rimegepant to healthy male subjects, 78% of the total radioactivity was recovered in faeces and 24% in urine. Unchanged rimegepant is the major single component in excreted faeces (42%) and urine (51%).
Linearity/non-linearity.
Rimegepant exhibits greater than dose proportional increases in exposure following single oral administration, which appears to be related to a dose-dependent increase in bioavailability.
Age, sex, weight, race, ethnicity.
No clinically significant differences in the pharmacokinetics of rimegepant were observed based on age, sex, race/ethnicity, body weight, migraine status, or CYP2C9 genotype.
Renal impairment.
In a dedicated clinical study comparing the pharmacokinetics of rimegepant in subjects with mild (estimated creatinine clearance [CLcr] 60-89 mL/min), moderate (CLcr 30-59 mL/min), and severe (CLcr 15-29 mL/min) renal impairment to that with normal subjects (healthy pooled control), a less than 50% increase in total rimegepant exposure was observed following a single 75 mg dose. The unbound AUC of rimegepant was 2.57-fold higher in subjects with severe renal impairment. Nurtec ODT has not been studied in patients with end-stage renal disease (CLcr < 15 mL/min).
Hepatic impairment.
In a dedicated clinical study comparing the pharmacokinetics of rimegepant in subjects with mild, moderate, and severe hepatic impairment to that of normal subjects (healthy matched control), the exposure of rimegepant (unbound AUC) following a single 75 mg dose was 3.89-fold higher in subjects with severe impairment (Child-Pugh class C). There were no clinically meaningful differences in the exposure of rimegepant in subjects with mild (Child-Pugh class A) and moderate hepatic impairment (Child-Pugh class B) compared to subjects with normal hepatic function.
5.3 Preclinical Safety Data
Genotoxicity.
Rimegepant was negative in in vitro (reverse mutation in bacterial cells and cytogenetics in Chinese Hamster Ovary [CHO] cells) and in vivo (rat oral micronucleus) assays.
Carcinogenicity.
Oral administration of rimegepant to Tg.rasH2 mice (up to 300 mg/kg/day) for 26 weeks, and to rats for 91-100 weeks (up to 45 mg/kg/day), resulted in no evidence of drug-induced tumours in either species. In these animal studies, the plasma exposure (AUC) at the highest dose tested was approximately 308x (mice) and 24 to 40x (rats, males and females respectively) the human AUC at 75 mg/day.6 Pharmaceutical Particulars
6.1 List of Excipients
Gelatin (sourced from fish); mannitol (E421); mint flavour; sucralose.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C. Keep in the original packaging to protect from moisture.
6.5 Nature and Contents of Container
Blisters made of polyvinyl chloride (PVC), oriented polyamide (OPA) and aluminium foil and sealed with a peelable aluminium foil.
Available in blister packs of 2, 4, 8 or 16 orally disintegrating tablets.
Not all pack sizes may be marketed.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
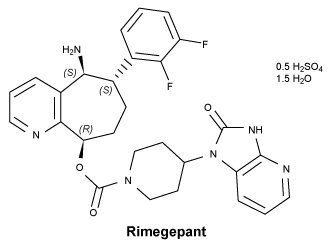
CAS number.
1289023-67-1.7 Medicine Schedule (Poisons Standard)
Schedule 4.
Summary Table of Changes
