What is in this leaflet
This leaflet answers some common questions about OCREVUS. It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given OCREVUS against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
What OCREVUS is used for
OCREVUS contains an active ingredient called ocrelizumab.
OCREVUS is used to treat relapsing forms of multiple sclerosis (RMS) and primary progressive multiple sclerosis (PPMS).
OCREVUS decreases the number of flare-ups (relapses) and slows the worsening of RMS.
OCREVUS also slows the worsening of PPMS.
In MS (multiple sclerosis) your immune system mistakenly attacks the protective layer (myelin) around your nerve cells. This causes inflammation and damage which stops your nervous system working properly.
OCREVUS works on your immune system so that it may reduce the inflammation and attacks on your nervous system reducing the chance of a flare-up and slowing the worsening of your disease.
Your doctor, however, may have prescribed OCREVUS for another purpose.
Ask your doctor if you have any questions about why OCREVUS has been prescribed for you.
This medicine is available only with a doctor's prescription.
This medicine is not addictive.
Before you are given OCREVUS
If you are not sure whether you should start taking this medicine, talk to your doctor.
When you must not be given OCREVUS
Do not use OCREVUS:
- if you have had an allergic reaction to OCREVUS or any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not use Ocrevus if the package is torn, shows signs of tampering or if the expiry date printed on packaging has passed. If you take this medicine after the expiry date has passed it may not work as well.
Before you are given OCREVUS
Tell your doctor if:
- you have an infection, or a history of a recurring or long-term infection such as Hepatitis B
If you are taking or have taken medicines which affect your immune system, you may have an increased risk of infections. - you are taking or have previously taken medicines which may affect your immune system, such as other medicines for MS
You should not receive OCREVUS if you are taking other medicines which may affect your immune system, such as other medicines for MS, as this may increase your risk of developing an infection.
Your doctor will consider the best time for you to begin treatment with OCREVUS.
There have been reports of a rare, serious brain infection called PML (progressive multifocal leuco-encephalopathy) in patients receiving medicines for MS. PML can cause severe disability or even death.
Symptoms of PML can be similar to those of MS. Tell your partner or carer about your OCREVUS treatment. They might notice symptoms that you do not, such as changes in movement or behaviour, which your doctor may need to investigate. - you intend to have or have had immunisation with any vaccine
Some vaccines should not be given at the same time as OCREVUS or in the months after you receive OCREVUS. Your doctor will check if you should have any vaccines before you receive OCREVUS.
OCREVUS may affect your normal response to a vaccine. - you have cancer or have had cancer in the past
Medicines that work on the immune system may increase the risk of some cancers. - you are allergic to any other medicines or any other substances such as foods, preservatives or dyes
- you are pregnant or intend to become pregnant
It is not known whether OCREVUS is harmful to an unborn baby. It is not recommended that you are given OCREVUS while you are pregnant.
If you are of child bearing potential, it is recommended that you do not become pregnant for 6 months following the end of treatment with OCREVUS.
If you are of child bearing potential, it is recommended that you use effective contraceptive methods during treatment and for up to 6 months following the end of treatment with OCREVUS. - you are breast feeding or plan to breast feed
It is not known if OCREVUS passes into breast milk. It is recommended that you discontinue breast feeding while you are treated with OCREVUS.
If you have not told your doctor about any of the above, tell them before you start taking OCREVUS.
Use in children
The safety and effectiveness of OCREVUS in children and adolescents under 18 years of age have not been established.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you have bought without a prescription from a pharmacy, supermarket or health food shop. As well as OCREVUS, there are other treatments (including those for MS, or to treat other conditions) which could affect your immune system and so could affect your ability to fight infections. If you have used another MS treatment in the past, your doctor may ask you to stop the other medicine in advance of starting treatment with OCREVUS.
As OCREVUS may cause a temporary drop in your blood pressure, your doctor may advise you to temporarily stop taking your blood pressure medicine before you are given OCREVUS.
How OCREVUS is given
Follow all directions given to you by your doctor or nurse carefully. They may differ from the information contained in this leaflet.
OCREVUS must be prepared by a healthcare professional and will be given in a hospital or clinic by a doctor or nurse.
OCREVUS is given by slow infusion into a vein (intravenous (IV) infusion).
The amount of OCREVUS you are given and how long each infusion lasts are different for the first and following doses. Each infusion is given on a different day.
Before you receive OCREVUS you will be given other medicines to help reduce the severity of possible infusion reactions.
The first infusion: you will be given 300 mg of OCREVUS by IV infusion over about 2.5 hours.
The second infusion: if the first infusion was well tolerated, you will be given 300 mg of OCREVUS by IV infusion 2 weeks after the first infusion.
Subsequent infusions: if the previous infusion was well tolerated, you will be given 600 mg of OCREVUS by IV infusion 6 months after the previous infusion. The infusion will be over about 3.5 hours.
If you did not experience a serious infusion related reaction with any previous infusion, a shorter infusion of approximately 2 hours can be given for subsequent doses.
You will be closely monitored during each infusion. Your doctor may adjust your infusion depending on how well each one is tolerated.
If you miss a dose
As OCREVUS is given under the supervision of your doctor, you are unlikely to miss a dose. However, if you forget or miss your appointment to receive OCREVUS, you should not wait until the next planned dose but make another appointment as soon as possible.
If you take too much (overdose)
As OCREVUS is given under the supervision of your doctor, it is very unlikely that you will be given too much. However, if you experience any side effects after being given OCREVUS, tell your doctor immediately.
While you are receiving OCREVUS
Things you must do
Tell your doctor or nurse immediately if you have any signs or symptoms of an infusion reaction or allergic reaction, or heart problems.
Some signs and symptoms can include:
- swelling of your face, lips, tongue or throat with difficulty breathing
- swelling of other parts of your body
- shortness of breath, wheezing or trouble breathing
- rash, itching or hives on the skin
- feeling sick (nausea)
- fever, chills
- feeling tired
- headache
- chest pain
- abnormal or irregular heartbeat
Tell your doctor or nurse straight away if you have any signs of infection during or after Ocrevus treatment. Tell your doctor even if it has been some time since your infusion. Some signs and symptoms can include:
- fever or chills
- cough that does not go away
- herpes (such as cold sores, shingles or genital sores)
Your doctor may need to do a blood test to check the level of your white blood cells.
Tell your partner or caregiver you are receiving OCREVUS and ask them to tell you if they notice any changes in your movement or behaviour. If they notice any changes you should tell your doctor about them immediately. Your doctor may need to perform some tests and alter your treatment.
Tell all doctors, dentists and pharmacists who are treating you that you are receiving OCREVUS.
Tell your doctor that you are receiving OCREVUS before any vaccinations.
Tell your doctor if you become pregnant or intend to start a family while receiving OCREVUS, or if you intend to breast feed whilst receiving OCREVUS.
If you become pregnant or plan to become pregnant, speak to the doctor about vaccinations for your baby.
Tell your baby’s doctor about your treatment with OCREVUS so that they can decide when your baby should be vaccinated. Be sure to keep all of your appointments with your doctor so that your progress can be checked.
Things you must not do
Do not stop your OCREVUS treatment without talking to your doctor first.
Tell your doctor if you feel that OCREVUS is not helping your condition.
Do not take any other medicines, whether they require a prescription or not without first telling your doctor or consulting with a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how OCREVUS affects you.
Side effects
Tell your doctor or nurse as soon as possible if you do not feel well while you are receiving OCREVUS.
OCREVUS helps most people with multiple sclerosis, but it may have unwanted side effects in some people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
During an infusion
Tell your doctor or nurse immediately if you notice any of the following while receiving an infusion:
- swelling of your face, lips, tongue or throat with difficulty breathing
- swelling of other parts of your body
- shortness of breath, wheezing or trouble breathing
- rash, itching or hives on the skin
- feeling sick (nausea)
- fever, flushing or chills
- cough, throat irritation or pain
- feeling tired
- headache
- dizziness or light headedness
- fast heartbeat
These may be serious side effects. You may need medical attention.
After an infusion
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- swelling of your face, lips, tongue or throat with difficulty breathing
- swelling of other parts of your body
- shortness of breath, wheezing or trouble breathing
- skin problems including rash, itchiness or hives
- fever, flushing or chills
- cough, throat irritation or pain
- feeling tired
- headache
- dizziness or light headedness
- fast heartbeat
These may be serious side effects. You may need medical attention.
Tell your doctor or pharmacist if you notice any of the following:
- signs of an infection such as fever or chills
- cold sore, shingles or genital sores
- stuffy nose or chest
- thick mucus in the nose, throat or chest
- persistent cough
- itchy, red or sore eyes
- diarrhoea, vomiting and/or abdominal pain
- skin infection (cellulitis)
- sinus infections
Tell your doctor if you experience signs and symptoms of infection, even if it has been some time (e.g. several weeks or months) since your last infusion. Your doctor may need to do a blood test to check your white blood cell levels.
This is not a complete list of all possible side effects. Your doctor or pharmacist has a more complete list. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell, even if it is not on this list.
Ask your doctor, nurse or pharmacist if you don't understand anything in this list.
Product description
Storage
Store OCREVUS at 2°C to 8°C (Refrigerate. Do not freeze). Protect from light.
Availability
OCREVUS is supplied as a single-dose glass vial containing 10 mL of solution for intravenous infusion (30 mg/mL). It is diluted before infusion into a vein.
What OCREVUS looks like
OCREVUS is a clear or slightly opalescent, colourless to pale brown liquid.
Ingredients
Each vial of OCREVUS contains 300 mg of the active ingredient ocrelizumab. It also contains:
- sodium acetate trihydrate
- trehalose dihydrate
- acetic acid - glacial
- polysorbate 20
- water for injections
Distributor
Distributor
OCREVUS is distributed by:
Roche Products Pty Limited
ABN 70 000 132 865
Level 8, 30 – 34 Hickson Road
Sydney NSW 2000
AUSTRALIA
Medical enquiries: 1800 233 950
Please check with your pharmacist for the latest Consumer Medicine Information (CMI).
Australian Registration Number:
AUST R 275778
This leaflet was prepared on 10 May 2021
Published by MIMS July 2021





 Results of the pre-specified pooled analyses of time to CDP sustained for at least 12 weeks (40% risk reduction for Ocrevus compared to interferon beta-1a, p = 0.0006) were highly consistent with the results sustained for at least 24 weeks (40% risk reduction for Ocrevus compared to interferon beta-1a, p = 0.0025).
Results of the pre-specified pooled analyses of time to CDP sustained for at least 12 weeks (40% risk reduction for Ocrevus compared to interferon beta-1a, p = 0.0006) were highly consistent with the results sustained for at least 24 weeks (40% risk reduction for Ocrevus compared to interferon beta-1a, p = 0.0025). Key clinical and MRI efficacy results are presented in Table 7 and Figure 2.
Key clinical and MRI efficacy results are presented in Table 7 and Figure 2.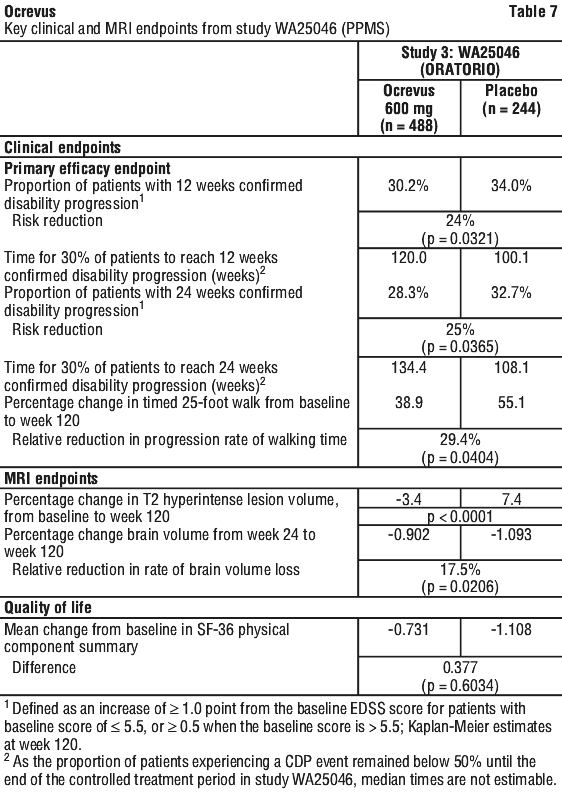
 A post-hoc analysis suggested that patients who are 50 years of age or below, or patients who have inflammation determined by MRI (Gd enhancing or T2 lesion) may receive a greater treatment benefit than patients who are over 50 years of age or patients who do not have inflammation by MRI.
A post-hoc analysis suggested that patients who are 50 years of age or below, or patients who have inflammation determined by MRI (Gd enhancing or T2 lesion) may receive a greater treatment benefit than patients who are over 50 years of age or patients who do not have inflammation by MRI.
