What is in this leaflet
This leaflet answers some common questions about OLUMIANT. It does not contain all the available information. It should not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking OLUMIANT against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, talk to your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What OLUMIANT is used for
OLUMIANT is used in adults as a treatment for moderate to severe:
- rheumatoid arthritis
- atopic dermatitis, also known as atopic eczema.
It can be used alone or with other medicines used to treat rheumatoid arthritis or with eczema medicines that you apply to the skin.
Rheumatoid arthritis is an autoimmune disease which means the body's immune system mistakenly attacks its own tissues. Symptoms include joint pain, tenderness, swelling and stiffness.
Atopic dermatitis is a chronic skin condition marked by intense itching, inflammation and dryness of skin.
OLUMIANT contains the active substance baricitinib. It belongs to a group of medicines called Janus Kinase (JAK) inhibitors.
OLUMIANT works by reducing the activity of an enzyme in the body called Janus Kinase, which is involved in inflammation. By reducing the activity of this enzyme, OLUMIANT helps:
- block parts of your immune system involved in rheumatoid arthritis and atopic dermatitis.
- improve the condition of your skin inflammation and reduce itching associated with atopic dermatitis.
Ask your doctor if you have any questions about why OLUMIANT has been prescribed for you. Your doctor may have prescribed it for another reason.
OLUMIANT is available only with a doctor's prescription.
OLUMIANT is not intended for use by children under the age of 18 years.
Before you take OLUMIANT
When you must not take it
Do not take OLUMIANT if you are taking any biological (injectable) treatment for your condition.
Do not take OLUMIANT if you have an allergy to:
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine after the expiry date printed on the pack. If it has expired, return it to your pharmacist for disposal.
Do not take this medicine if the packaging is torn or if the seals over the carton ends are missing or broken. If it is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking OLUMIANT, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have ever had any of the following medical conditions:
- are being treated for an infection, get a lot of infections or have infections that keep coming back
- tuberculosis or have been in close contact with someone who has tuberculosis
- diabetes
- hepatitis B or C
- shingles
- low white or red blood cells
- recently been vaccinated or plan to get vaccinated
- kidney problems
- high cholesterol
- liver problems
- cancer
- blood clots in the blood vessels of your legs (deep vein thrombosis) or lungs (pulmonary embolism).
Tell your doctor if you are pregnant or planning to become pregnant. It is not known if OLUMIANT will harm your unborn baby. You should use an effective method of contraception to avoid becoming pregnant while taking OLUMIANT and for at least one week after your final dose. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you are breastfeeding or plan to breast feed. It is not known if OLUMIANT passes into your breast milk. You should not breast feed if you are taking OLUMIANT.
Tell your doctor if you have ever had shingles (herpes zoster virus). OLUMIANT can reactivate shingles in people who have had shingles in the past.
Tell your doctor if you are a carrier of the hepatitis B virus (HBV) or hepatitis C virus (HCV), or if you have hepatitis B or hepatitis C infection. If you have the HBV or HCV in your blood, it may become active while you are taking OLUMIANT. This effect has been reported with other medicines used to treat rheumatoid arthritis.
Make sure you are up to date with all vaccinations before starting OLUMIANT.
Tell your doctor if you are scheduled for any vaccines or have recently been vaccinated. Some vaccines should not be given while you are taking OLUMIANT. Check with your doctor before receiving any vaccines.
If you have not told your doctor or pharmacist about any of the above, tell them before you start taking OLUMIANT.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may be affected by OLUMIANT or may affect how it works. These include:
- medications used to treat gout such as probenecid.
You may need different amounts of your medicines or you may need to take different medicines.
Tell your doctor or pharmacist if you are already using another medicine for your condition.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take OLUMIANT
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the pack, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how much you need to take each day.
OLUMIANT is supplied as 4 mg and 2 mg tablets.
The usual dose for rheumatoid arthritis is one 4 mg tablet taken once a day. Your doctor may reduce your dose once your condition is under control.
The usual dose for atopic dermatitis is 2 mg once a day. Your doctor may increase your dose to 4 mg once a day if you have not achieved sustained control of your condition with 2 mg and then may reduce your dose to 2 mg once a day once you have achieved sustained control of your condition with 4 mg once a day.
Your doctor may also adjust your dose depending on your condition, for example if you have a kidney problem, or are taking medication to treat gout.
How to take it
Swallow the tablet whole with a glass of water.
When to take it
Take OLUMIANT tablets at about the same time every day. Taking it at the same time every day will help you remember when to take it.
OLUMIANT can be taken with or without food.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition. It is important to keep taking your medicine even if you feel well.
If you forget to take it
If it is close to your next dose, skip the dose you missed and take your next dose when you are meant to. Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for missed tablets.
If you are not sure what to do, ask your doctor or pharmacist.
If you take too much (overdose)
Immediately telephone your doctor or the Australian Poisons Information Centre (telephone 13 11 26), or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much OLUMIANT. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention
While you are taking OLUMIANT
Things you must do
Tell your doctor if you experience signs and symptoms of infection such as:
- fever, sweating or chills
- muscle aches
- cough
- shortness of breath
- weight loss
- warm, red or painful skin or sores on your body
- burning when you urinate or urinating more often than normal
- feeling very tired.
OLUMIANT can lower the ability of your body to fight infections.
Tell your doctor if you get a painful swollen leg, chest pain, or shortness of breath as these can be signs of blood clots. Blood clots in the blood vessels of the legs (deep vein thrombosis) or lungs (pulmonary embolism) have been reported in patients taking OLUMIANT.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking OLUMIANT.
Tell any other doctors, dentists and pharmacists who treat you that you are taking OLUMIANT.
You should use an effective method of contraception to avoid becoming pregnant while taking OLUMIANT and for at least one week after your final dose. Your doctor can discuss with you the risks and benefits involved.
If you become pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked.
Your doctors will perform blood tests at regular intervals during your treatment to monitor your blood cells, liver enzymes and cholesterol.
Things you must not do
Do not take OLUMIANT to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine, or change the dosage, without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how OLUMIANT affects you.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking OLUMIANT.
OLUMIANT helps people with moderate to severe rheumatoid arthritis and atopic dermatitis, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
It can be difficult to tell whether side effects are the result of taking OLUMIANT, effects of your condition or side effects of other medicines you may be taking. For this reason it is important to tell your doctor of any change in your condition.
Do not be alarmed by the following list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor immediately or go to the Emergency Department at your nearest hospital if you notice any of the following:
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- severe rash, itching or hives
The above may be signs of an allergic reaction and may require urgent medical attention or hospitalisation.
Tell your doctor as soon as possible if you notice any of the following as they may be serious side effects and require medical attention:
- signs of an infection, such as fever, sweating or chills; muscle aches; cough; shortness of breath; weight loss; warm, red or painful skin or sores on your body; burning when you urinate or urinating more often than normal; feeling very tired.
- Signs of blood clots in the blood vessels of the legs (deep vein thrombosis) or lungs (pulmonary embolism) such as painful swollen leg, chest pain or shortness of breath.
- symptoms of shingles, such as headache; sensitivity to light; tingling, itching or painful skin rash with blisters.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- a cold, sore throat, runny or blocked nose; cough (respiratory tract infection)
- nausea (feeling sick)
- cold sore blisters
- acne (pimples)
- rash
- abdominal pain
- headache
The above list includes the more common side effects of your medicine.
Some side effects (for example, changes in cholesterol, liver or other enzymes, and blood cell levels) can only be found when your doctor does tests from time to time to check your progress.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
This is not a complete list of all possible side effects. Others may occur in some people and there may be side effects not yet known.
Ask your doctor or pharmacist if you don't understand anything on this list.
After taking OLUMIANT
Storage
Keep your tablets in the original pack until it is time to take them. If you take the tablets out of the pack they may not keep as well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store OLUMIANT or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep your medicine where children cannot reach. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product Description
What it looks like
OLUMIANT 4 mg tablets are pink, round, film-coated tablets, marked with "Lilly" on one side and "4" on the other side.

OLUMIANT 2 mg tablets are pale pink, oblong, film-coated tablets, marked with "Lilly" on one side and "2" on the other side.

Both tablets contain a dent on each face of the tablet to help you with picking them up.
Ingredients
Active Ingredient
- baricitinib 4 mg or 2 mg
Other Ingredients
- croscarmellose sodium
- magnesium stearate
- mannitol
- microcrystalline cellulose
- iron oxide red
- lecithin
- macrogol 3350
- polyvinyl alcohol
- purified talc
- titanium dioxide
This medicine does not contain sucrose, gluten, tartrazine or any other azo dyes.
Supplier
OLUMIANT is a product of:
Eli Lilly Australia Pty Limited
112 Wharf Road
West Ryde, NSW 2114
AUSTRALIA
®= Registered Trademark
If you have any questions about OLUMIANT, contact Eli Lilly at 1800 454 559 (Australia) or your healthcare professional for assistance.
Australian Registration Numbers
OLUMIANT 4 mg tablet - AUST R 277917
OLUMIANT 2 mg tablet - AUST R 277905
Further Information
This leaflet was prepared in February 2021
vA-02
Published by MIMS March 2021




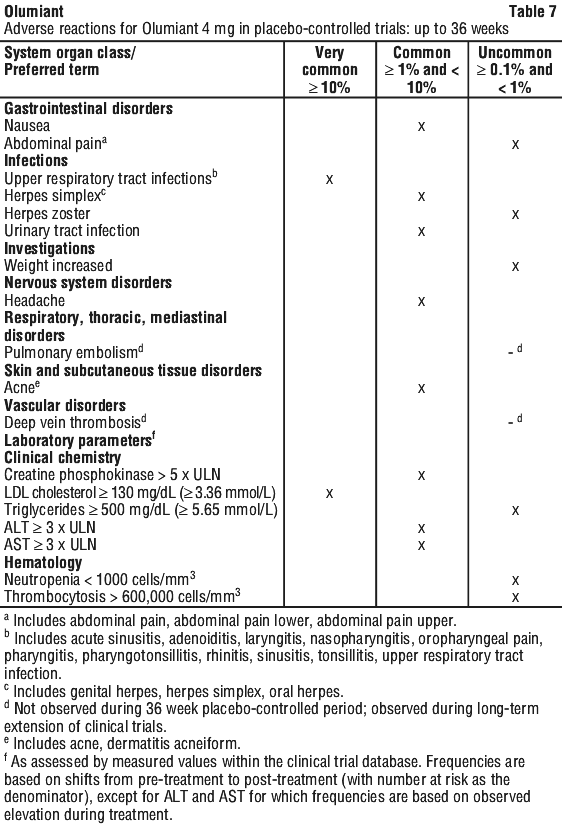
 In patients treated with any dose of baricitinib, adverse events that occurred in fewer than 1% of patients include myocardial infarction and B cell lymphoma.
In patients treated with any dose of baricitinib, adverse events that occurred in fewer than 1% of patients include myocardial infarction and B cell lymphoma.






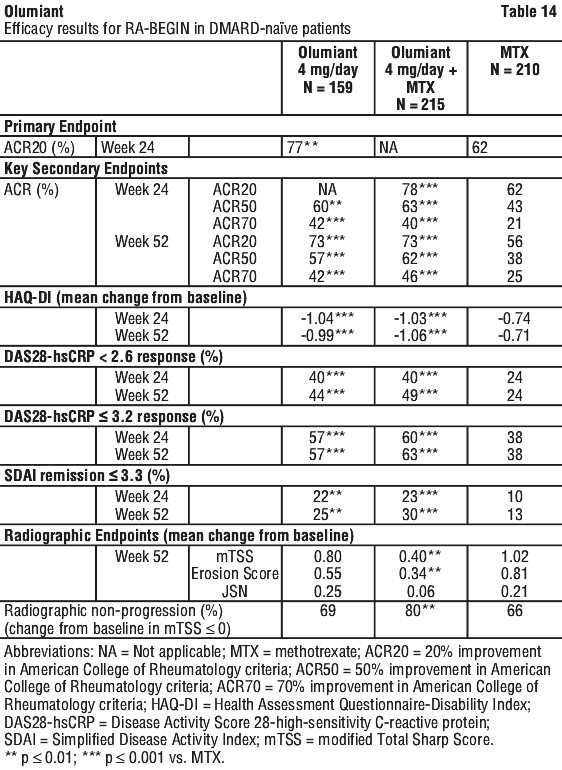
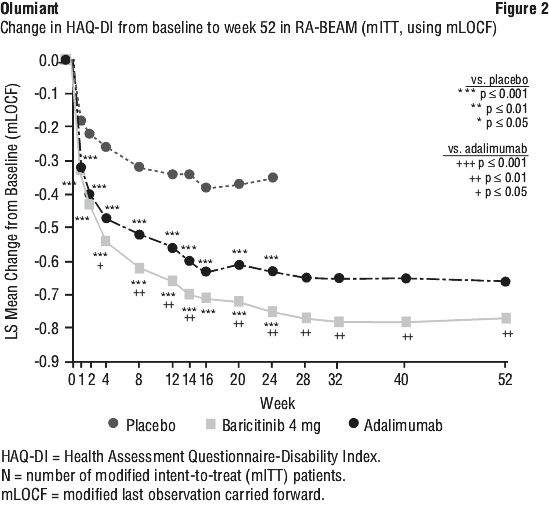 Treatment with Olumiant 4 mg, alone or in combination with cDMARDs, resulted in a significant improvement in pain compared to all comparators (placebo, MTX, and adalimumab), as measured on a 0-100 visual analogue scale, at 12 weeks. Greater pain reduction was seen as early as Week 1 and, in RA-BEGIN and RA-BEAM, this was maintained for up to 52 weeks.
Treatment with Olumiant 4 mg, alone or in combination with cDMARDs, resulted in a significant improvement in pain compared to all comparators (placebo, MTX, and adalimumab), as measured on a 0-100 visual analogue scale, at 12 weeks. Greater pain reduction was seen as early as Week 1 and, in RA-BEGIN and RA-BEAM, this was maintained for up to 52 weeks.