1 Name of Medicine
Omalizumab (rch).
Omlyclo (omalizumab) is a biosimilar medicine to Xolair (omalizumab).
The evidence for comparability supports the use of Omlyclo for the listed indications.
2 Qualitative and Quantitative Composition
Omalizumab is a recombinant DNA-derived humanised monoclonal antibody produced in Chinese hamster ovary cells that selectively binds to human immunoglobulin E (IgE).
Solution for injection in pre-filled syringe with needle guard.
Each pre-filled syringe of 0.5 mL contains 75 mg of omalizumab.
Each pre-filled syringe of 1 mL contains 150 mg of omalizumab.
Solution for injection in pre-filled pen.
Each pre-filled pen of 0.5 mL contains 75 mg of omalizumab. Each pre-filled pen of 1 mL contains 150 mg of omalizumab.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Solution for injection in pre-filled syringe and pre-filled pen.
Clear to slightly opalescent, colourless to pale brownish-yellow preservative free solution in a pre-filled syringe or pre-filled pen.4.1 Therapeutic Indications
Allergic asthma.
Children 6 to < 12 years of age.
In children aged 6 to < 12 years, Omlyclo is indicated as add-on therapy to improve asthma control in patients with severe allergic asthma who have documented exacerbations despite daily high dose inhaled corticosteroids, and who have immunoglobulin E levels corresponding to the recommended dose range (see Section 4.2 Dose and Method of Administration, Table 1).
Adults and adolescents ≥ 12 years of age.
Omlyclo is indicated for the management of adult and adolescent patients with moderate to severe allergic asthma, who are already being treated with inhaled steroids, and who have serum immunoglobulin E levels corresponding to the recommended dose range (see Section 4.2 Dose and Method of Administration, Table 1).
Chronic rhinosinusitis with nasal polyps (CRSwNP).
Omlyclo is indicated as add-on treatment in adult patients (18 years of age and above) for the treatment of severe CRSwNP with inadequate response to intranasal corticosteroids.
Recommended dosing is determined by serum immunoglobulin E levels and body weight corresponding to the recommended dose range in the Product Information (see Section 4.2 Dose and Method of Administration).
Chronic spontaneous urticaria (CSU).
Omlyclo is indicated for adults and adolescents (12 years of age and above) with chronic spontaneous urticaria who remain symptomatic despite H1 antihistamine treatment.4.2 Dose and Method of Administration
Dosing principles for allergic asthma and CRSwNP.
Omlyclo treatment for chronic rhinosinusitis with nasal polyps (CRSwNP) should be initiated by specialists experienced in the diagnosis and treatment of CRSwNP.
Dosing for asthma and CRSwNP follows the same dosing principles. The appropriate dose and dosing frequency of Omlyclo for these conditions is determined by baseline immunoglobulin E (IgE) (IU/mL), measured before the start of treatment, and body weight (kg). Prior to initial dosing, patients should have their IgE level determined by any commercial serum total IgE assay for their dose assignment. Based on these measurements 75 to 600 mg of Omlyclo in 1 to 4 injections may be needed for each administration. See Table 1 for dose determination in allergic asthma and CRSwNP and for a dose conversion chart.
Measurement of serum IgE levels.
Any commercial serum total IgE assay may be used for determination of serum total IgE for initial dose assignment. However, Omlyclo can interfere with accurate quantitation of serum IgE levels. The total IgE levels while on active treatment of omalizumab increased an average of 4-fold postdose as a result of omalizumab-IgE binding. If it is necessary to measure serum total IgE in subjects currently on Omlyclo treatment or who have discontinued within the last 12 months, the Abbott IMX assay has been shown to demonstrate reliable serum total IgE measurements.
Treatment duration, monitoring and dose adjustments.
Doses do not need to be adjusted for variations in serum IgE over time. Data from clinical studies suggest that, in the absence of omalizumab treatment, there is no significant temporal variation in serum total IgE levels.
Omlyclo is intended for long-term treatment. In treatment for allergic asthma, at least 16 weeks of treatment is required to adequately assess whether or not a patient is responding to Omlyclo. The decision to continue Omlyclo should be based on whether a marked improvement in overall asthma control is seen (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Discontinuation of Omlyclo treatment generally results in a return to elevated free IgE levels and associated symptoms. Total IgE levels are elevated during treatment and remain elevated for up to one year after the discontinuation of treatment. Therefore, re-testing of IgE levels during Omlyclo treatment cannot be used as a guide for dose determination. Dose determination after treatment interruptions lasting less than one year should be based on serum IgE levels obtained at the initial dose determination. Total serum IgE levels may be re-tested for dose determination if treatment with Omlyclo has been interrupted for one year or more.
Doses should be adjusted for significant changes in body weight (see Table 1). No data are currently available to support dose adjustments based on changes in serum total IgE with increasing age.
Reduction of inhaled corticosteroids may be attempted after 16 weeks of treatment with Omlyclo in patients with stable, well-controlled asthma. The dose of corticosteroid should be reduced gradually under medical supervision. In some patients, inhaled corticosteroids can be tapered off completely. Omlyclo should not be abruptly substituted for inhaled corticosteroids.
In clinical trials for CRSwNP, changes in CRSwNP score (NPS) and nasal congestion score (NCS) were observed as early as the first assessment at 4 weeks. The need for continued therapy should be periodically reassessed based upon the patient's disease severity and level of symptom control.
Dosage for allergic asthma and CRSwNP.
Omlyclo is administered subcutaneously every two or four weeks according to the dose determination chart (Table 1). Doses (mg) and dosing frequency are determined by baseline serum total IgE level (IU/mL), measured before the start of treatment, and body weight (kg).
 There is limited data in patients whose baseline IgE levels or body weight in kg are outside the limits of the dosing tables. Doses greater than 750 mg were not studied in allergic asthma in the pivotal clinical studies and are not recommended. There is limited data on doses greater than 750 mg from the pivotal studies in CRSwNP and therefore higher doses are not recommended.
There is limited data in patients whose baseline IgE levels or body weight in kg are outside the limits of the dosing tables. Doses greater than 750 mg were not studied in allergic asthma in the pivotal clinical studies and are not recommended. There is limited data on doses greater than 750 mg from the pivotal studies in CRSwNP and therefore higher doses are not recommended.
Patients with severe asthma and a baseline IgE lower than 76 IU/mL were less likely to experience benefit (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Prescribing physicians should ensure that adult and adolescent patients with IgE below 76 IU/mL and children (6 to < 12 years of age) with IgE below 200 IU/mL have unequivocal in vitro reactivity (RAST) to a perennial allergen before starting therapy.
The prescribing of Omlyclo in children with asthma should be done in conjunction with a paediatrician, respiratory physician or immunologist.
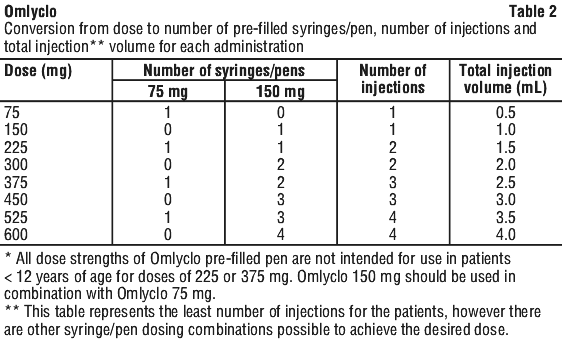 For doses of 225 or 375 mg. Omlyclo 150 mg should be used in combination with Omlyclo 75 mg.
For doses of 225 or 375 mg. Omlyclo 150 mg should be used in combination with Omlyclo 75 mg.
Dosage for chronic spontaneous urticaria (CSU).
The recommended dose is 300 mg by subcutaneous injection every four weeks. Some patients may achieve control of their symptoms with a dose of 150 mg every four weeks.
Prescribers are advised to periodically reassess the need for continued therapy.
Clinical trial experience of long-term treatment beyond 6 months in this indication is limited.
Omlyclo should be used as add-on therapy to H1 antihistamine treatment.
Method of administration.
For subcutaneous administration only. Omlyclo must not be administered by the intravenous or intramuscular route.
Doses of more than 150 mg should be divided across two or more injection sites.
Pre-filled syringe and pre-filled pen solution for injection.
All dose strengths of Omlyclo pre-filled pen are not intended for use in patients < 12 years of age. Omlyclo 75 mg pre-filled syringe and Omlyclo 150 mg pre-filled syringe, may be used in children 6 to 11 years of age with allergic asthma.
If more than one injection is needed to achieve the required dose, injections should be divided across two or more injection sites (see Table 2).
After proper training in subcutaneous injection technique, patients or the caregiver may self-inject Omlyclo if a physician determines that it is appropriate. It is recommended that the first 3 doses be administered by or under the supervision of a healthcare professional.
Patients or caregivers should be instructed to inject the full amount of Omlyclo according to the "Instructions for use" at the end of the leaflet.
Product is for single use in one patient only. Discard any residue.4.3 Contraindications
Hypersensitivity to omalizumab or any other component of the formulation.
4.4 Special Warnings and Precautions for Use
Abrupt discontinuation of systemic or inhaled corticosteroids after initiation of omalizumab therapy in allergic asthma or CRSwNP is not recommended. Decreases in corticosteroids should be performed under the direct supervision of a physician and may need to be performed gradually.
Allergic reactions.
As with any protein, local or systemic allergic reactions, including anaphylaxis, may occur. Anaphylactic reactions were rare in clinical trials (see Section 4.8 Adverse Effects (Undesirable Effects)). In post-marketing experience, anaphylaxis and anaphylactoid reactions have been reported following the first and subsequent administrations of omalizumab. Most of these reactions occurred within 2 hours after omalizumab administration, some occurred beyond 2 hours and even beyond 24 hours after injections. The majority of anaphylactic reactions occurred within the first 3 doses of omalizumab. A history of anaphylaxis unrelated to omalizumab may be a risk factor for anaphylaxis following omalizumab administration. It is recommended that patients with a known history of anaphylaxis have each dose of omalizumab administered in a setting where there is treatment for anaphylaxis available. Patients should be informed that such reactions are possible and prompt medical attention should be sought if allergic reactions occur.
Serum sickness and serum sickness-like reactions, which are delayed allergic type III reactions, have rarely been seen in patients treated with humanized monoclonal antibodies including omalizumab. The onset has typically been 1-5 days after administration of the first or subsequent injections, also after long duration of treatment. Symptoms suggestive of serum sickness include arthritis/arthralgia, rash (urticaria or other forms), fever and lymphadenopathy. Patients should be advised to report any suspected symptoms.
Churg-Strauss syndrome and hypereosinophilic syndrome.
Patients with severe asthma may rarely present systemic hypereosinophilic syndrome or allergic eosinophilic granulomatous vasculitis (Churg-Strauss syndrome), both of which are usually treated with systemic corticosteroids. In rare cases, patients on therapy with anti-asthma agents, including omalizumab, may present or develop systemic eosinophilia and vasculitis. A causal association between omalizumab and these underlying conditions has not been established. These events are commonly associated with the reduction of oral corticosteroid therapy. In these patients, physicians should be alert to the development of marked eosinophilia, vasculitic rash, worsening pulmonary symptoms, paranasal sinus abnormalities, cardiac complications, and/or neuropathy. Discontinuation of omalizumab should be considered in all severe cases with the above-mentioned immune system disorders.
Immunogenicity.
As with all protein pharmaceuticals, a small percentage of patients may potentially develop antibodies to the protein (see Section 4.8 Adverse Effects (Undesirable Effects)).
Other IgE-associated disorders.
Omalizumab has not been studied in patients with anaphylaxis, hyperimmunoglobulin E syndrome, allergic bronchopulmonary aspergillosis, food allergy or atopic dermatitis. Parasitic infestation may also result in elevation of serum IgE concentrations. In a study of asthmatic patients who had been treated for gut parasites, the level of reinfection did not differ significantly between omalizumab and placebo groups and there were no serious or severe infections. There is no current evidence to suggest that parasitic infections are predisposed to by omalizumab.
Interpretation of serum IgE levels, skin patch and skin prick testing.
Patients treated with omalizumab have a rapid reduction in free IgE in the serum but an overall increase in the total serum IgE (commonly measured in clinical laboratories), which reflects free IgE and IgE bound by omalizumab. IgE measured following treatment cannot be used to guide treatment or dosing decisions.
Because omalizumab reduces free IgE in the serum and tissues, results of skin prick testing, patch testing, and RAST testing for hypersensitivity to potential allergens may be affected. A positive test to a potential allergen in a patient receiving omalizumab can be correctly interpreted as representing hypersensitivity to that allergen; however, a negative test in a patient receiving omalizumab may not be interpretable. Physicians are urged to use caution in interpreting such tests in patients receiving omalizumab.
Thrombocytopenia.
At serum concentrations in excess of maximum human exposure used in pivotal clinical trials, dose-related thrombocytopenia occurred in 2 out of 4 non-human primate species studied. The thrombocytopenia was more pronounced in juvenile animals. No omalizumab-related thrombocytopenia has been observed in clinical trials, but it has been reported in the post-market setting.
Omalizumab should be used with caution in patients with thrombocytopenia and patients with a history of thrombocytopenia. It is recommended that patients have a platelet count before commencing therapy with omalizumab and then periodically during treatment with omalizumab.
Arteriothrombolic events (ATE).
In controlled clinical trials, interim and final analyses of an observational study, a numerical imbalance of ATE was observed. See Section 4.8 Adverse Effects (Undesirable Effects).
Use in hepatic impairment.
There have been no studies on the effect of impaired hepatic function on the pharmacokinetics of omalizumab. Omalizumab should be administered with caution in these patients.
Use in renal impairment.
There have been no studies on the effect of impaired renal function on the pharmacokinetics of omalizumab. Omalizumab should be administered with caution in these patients.
Use in the elderly.
There are limited data available on the use of omalizumab in patients older than 65 years but there is no evidence that elderly patients require a different dosage from younger adult patients.
Paediatric use.
In allergic asthma, there are currently insufficient safety data to support the use of omalizumab in children under the age of 6 years.
In CRSwNP, safety and efficacy in patients below the age of 18 years has not been established.
In chronic spontaneous urticaria, there are currently insufficient safety data to support the use of omalizumab in children under the age of 12 years.
Effects on laboratory tests.
See Section 4.8 Adverse Effects (Undesirable Effects), Thrombocytopenia.4.5 Interactions with Other Medicines and Other Forms of Interactions
In clinical studies, omalizumab was effectively and safely used in conjunction with inhaled corticosteroids, inhaled beta agonists and oral antihistamines. No formal drug interaction studies have been performed with omalizumab.
Chronic rhinosinusitis with nasal polyps (CRSwNP).
In clinical studies omalizumab was used in conjunction with intranasal mometasone spray per protocol. Other commonly used concomitant medications included other intranasal corticosteroids, bronchodilators, antihistamines, leukotriene receptor antagonists, adrenergics/sympathomimetics, and local nasal anesthetics. There was no indication that the safety of omalizumab was altered with these other commonly used CRSwNP medications.
Chronic spontaneous urticaria (CSU).
In clinical studies in CSU omalizumab was used in conjunction with antihistamines (anti-H1, anti-H2) and leukotriene receptor antagonists (LTRAs). In the phase III studies Q4881g and Q4882g all patients received H1 antihistamines in addition to omalizumab or placebo. In the phase III study Q4883g, all patients received one or more H1 antihistamine(s), and/or H2 antihistamines and/or LTRAs in addition to omalizumab or placebo. There was no evidence that the safety of omalizumab was altered when used with these medicinal products relative to its known safety profile in allergic asthma. In addition, a population pharmacokinetic analysis showed no relevant effect of H2 antihistamines and LTRAs on omalizumab pharmacokinetics (see Section 5.2 Pharmacokinetic Properties).4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no human fertility data for omalizumab. Studies in cynomolgus monkeys showed no adverse effects of omalizumab on fertility or general reproductive performance of males or females at weekly doses up to 75 mg/kg SC (yielding about 10 times the maximum anticipated clinical exposure in adult patients, based on serum AUC).
(Category B1)
Human IgG antibodies are known to cross the placental barrier; therefore, omalizumab may be transmitted from the mother to the developing fetus. There are no well-controlled clinical studies of omalizumab in pregnant women. A prospective pregnancy registry study (EXPECT) in 250 pregnant women with asthma exposed to omalizumab showed the prevalence of major congenital anomalies was similar (8.1% vs 8.9%) between EXPECT and disease matched (moderate and severe asthma) patients. There was an increased rate of low birth weight among registry infants compared to infants in the other cohort. This study cannot definitively establish risk due to methodological limitations of the study, including small sample size, non-randomised design and potential differences between the registry population and the comparator cohort.
Animal data.
Studies in cynomolgus monkeys do not indicate direct or indirect harmful effects with respect to pregnancy, embryonic/fetal development, parturition or neonatal growth at weekly doses up to 75 mg/kg SC (yielding about 10 times the maximum anticipated clinical exposure in adult patients, based on serum AUC).
If clinically needed, the use of omalizumab may be considered during pregnancy.
Immunoglobulins G (IgGs) are present in human milk and therefore it is expected that omalizumab will be present in human milk.
The EXPECT study, with 154 infants who had been exposed to omalizumab during pregnancy and through breast-feeding did not indicate adverse effects on the breast-fed infant. The interpretation of data may be impacted due to methodological limitations of the study, including small sample size and non-randomised design.
Given orally, immunoglobulin G proteins undergo intestinal proteolysis and have poor bioavailability. No effects on the breast-fed newborns/infants are anticipated. Consequently, if clinically needed, the use of omalizumab may be considered during breast-feeding.
Animal data.
In order to assess the effect of omalizumab on late gestation, and to evaluate the placental transfer and milk excretion of omalizumab, doses of 75 mg/kg/week were administered subcutaneously to female cynomolgus monkeys. Transport of omalizumab into maternal milk was limited. The serum levels of omalizumab observed in dams, fetuses, and neonates are consistent with reported transport and distribution of IgG class immunoglobulins.4.7 Effects on Ability to Drive and Use Machines
Patients receiving omalizumab should be informed that if they experience dizziness, fatigue, faintness or somnolence they should not drive or use machines.
4.8 Adverse Effects (Undesirable Effects)
Clinical trial experience in allergic asthma.
Adverse reactions with omalizumab were observed (all studies with adult and adolescent patients 12 years of age and older) at a frequency of 6.6% of patients, treated with active drug, during clinical trials.
The most commonly associated adverse drug reactions were injection site reactions, including injection site pain, swelling, itching and redness (1.7%) and headaches (1%).
Other adverse reactions most frequently observed were weight increase (0.7%), urticaria (0.4%), fatigue, arm swelling, nausea, pharyngitis and skin rashes (all at 0.3%). Most of these events were mild or moderate in severity.
In clinical trials with patients 6 to < 12 years of age, the most commonly reported adverse reactions were headache, pyrexia and upper abdominal pain. Most of the events were mild or moderate in severity.
The adverse reactions listed in Table 3 were recorded in clinical studies in the total allergic asthma safety population treated with omalizumab. Adverse reactions are ranked under headings of frequency using the following convention: common (> 1/100; < 1/10), uncommon (> 1/1000; < 1/100), rare (< 1/1000).
 The frequencies of adverse reactions in the active treatment group adult and adolescent patients were very similar to those observed in the placebo group. Weight increase (0.7% vs 0.2%, placebo), urticaria (0.4% vs 0.1%, placebo) and local injection site reactions (1.7% vs 1.3%, placebo) were slightly more commonly observed in the active treatment group than in placebo group patients.
The frequencies of adverse reactions in the active treatment group adult and adolescent patients were very similar to those observed in the placebo group. Weight increase (0.7% vs 0.2%, placebo), urticaria (0.4% vs 0.1%, placebo) and local injection site reactions (1.7% vs 1.3%, placebo) were slightly more commonly observed in the active treatment group than in placebo group patients.
Adverse events (AEs).
The most common adverse events observed (frequency of > 20%) in this adult and adolescent patient population, were headaches, viral infections and upper respiratory tract infections. The frequencies of all adverse events for both treated (N = 1763) and placebo group patients (N =1278) for all studies were very similar.
Serious adverse events (SAEs).
Serious adverse events were reported for 2.6% of omalizumab treated patients and 2.8% of placebo-treated patients. The most frequently reported SAE's were appendicitis and fractures (0.2% for both treatment groups and both events). Frequencies of all SAEs by body system were comparable for both treatment groups.
Allergic asthma (adult and adolescent study population).
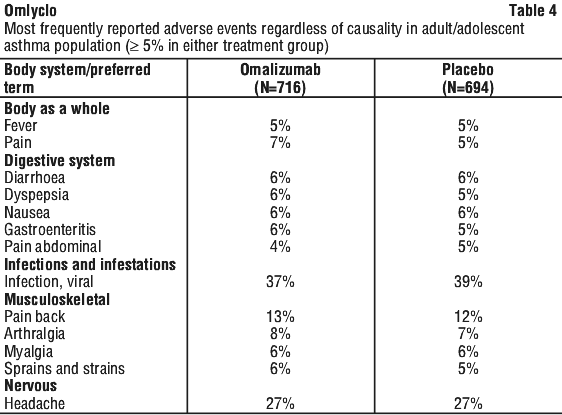
The most frequently observed adverse events in this population (N = 716 omalizumab patients, N = 694 placebo patients) were viral infections, upper respiratory tract infections, sinusitis and headaches (frequency ≥ 20%). Adverse reactions occurred in 5.6% of omalizumab-treated patients and in 5.2% of patients receiving placebo. The most frequently reported events of this type were headaches (1.3% vs 1.2%, placebo) and fatigue (0.6% vs 0%, placebo). All other suspected omalizumab drug related adverse events occurred with a frequency of < 0.5%.
Serious episodes of asthma related events requiring hospitalisation were observed in 0.2% of omalizumab-treated patients and 1.3% of placebo-treated patients, in the asthma studies.
The most frequently observed (≥ 5%) adverse events in studies with asthma patients 12 years of age and older are provided in Table 4.
Clinical trial experience in chronic rhinosinusitis with nasal polyps (CRSwNP).
Summary of the safety profile.
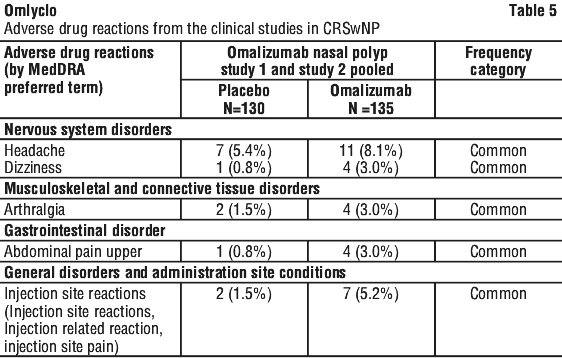
The data described below reflect data from two placebo-controlled studies in patients ≥ 18 years of age. In these studies, patients received either omalizumab 150 to 600 mg every 2 or 4 weeks or placebo. All patients received background intranasal mometasone therapy. The safety profile in patients with CRSwNP was consistent with that in allergic asthma and CSU. The most frequently (> 3%) reported adverse drug reactions, which were higher in frequency in comparison to placebo are shown in Table 5.
Tabulated summary of adverse drug reactions from the clinical studies.
Table 5 lists the adverse drug reactions recorded in clinical studies in the total nasal polyp safety population treated with omalizumab by system organ class and by frequency. Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).
Clinical trial experience in chronic spontaneous urticaria (CSU).
Chronic spontaneous urticaria (CSU). Summary of the safety profile.
The safety and tolerability of omalizumab were investigated with the doses of 75 mg, 150 mg and 300 mg every four weeks in 975 CSU patients, 242 of whom received placebo. 733 patients were treated with omalizumab for up to 12 weeks and 490 patients for up to 24 weeks. 175 and 412 patients were treated for up to 12 weeks and 87 and 333 patients were treated for up to 24 weeks at the recommended doses of 150 mg and 300 mg respectively.
During clinical studies with adult and adolescent patients (12 years of age and older) the most commonly reported adverse reactions observed were headache and nasopharyngitis.
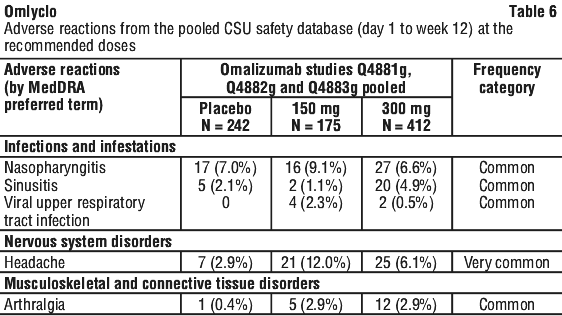
Tabulated summary of adverse reactions from the clinical studies at the recommended doses (150 mg and 300 mg).
Adverse reactions (events occurring in ≥ 1% of patients in any treatment group and ≥ 2% more frequently in any omalizumab treatment group than in the placebo group after medical review) reported at the recommended doses (150 mg and 300 mg) in the three pooled Phase III studies are listed by MedDRA system organ class (Table 6). Within each system organ class, the adverse reactions are ranked by frequency, with the most frequent reactions listed first. The corresponding frequency category for each adverse reaction is based on the following convention (CIOMS III): very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1000); very rare (< 1/10,000); not known (cannot be estimated from the available data).
 Additional events reported anytime during the day 1 to week 24 treatment period (studies Q4881g and Q4883g) that met the criteria of adverse reactions.
Additional events reported anytime during the day 1 to week 24 treatment period (studies Q4881g and Q4883g) that met the criteria of adverse reactions.
Infections and infestations.
Upper respiratory tract infections (placebo 3.1%, 150 mg 3.4%, 300 mg 5.7%), urinary tract infection (placebo 1.8%, 150 mg 4.6%, 300 mg 2.4%).
Nervous system disorders.
Sinus headache (placebo 0%, 150 mg 2.3%, 300 mg 0.3%).
Musculoskeletal and connective tissue disorders.
Myalgia (placebo 0%, 150 mg 2.3%, 300 mg 0.9%), pain in extremity (placebo 0%, 150 mg 3.4%, 300 mg 0.9%), musculoskeletal pain (placebo 0%, 150 mg 2.3%, 300 mg 0.9%).
General disorders and administration site conditions.
Pyrexia (placebo 1.2%, 150 mg 3.4%, 300 mg 0.9%).
Injection site reactions.
Injection site reactions occurred during the studies in more omalizumab-treated patients than placebo patients (2.7% at 300 mg, 0.6% at 150 mg, 0.8% with placebo). They included: swelling, erythema, pain, bruising, itching, bleeding and urticaria.
Comparability of Omlyclo and Xolair - adverse events.
There were no notable differences in the incidence or nature of Adverse Events (AEs) between the CT-P39 and Xolair treatment groups across Studies CT-P39 3.1 and CT-P39 1.1, and the safety profile of each treatment group was in line with the known safety profile of Xolair. As a result, the analysis of AEs obtained in the clinical studies with CT-P39 illustrated that the benefit-risk profile of CT-P39 is similar to that of Xolair.
Post-marketing observations.
The following reactions have been identified through spontaneous reporting.
Immune system disorders.
Anaphylaxis and anaphylactoid reactions have been reported following the first or subsequent administrations, serum sickness (see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects), Allergic events).
Skin and subcutaneous disorders.
Alopecia.
Blood and lymphatic system disorders.
Idiopathic severe thrombocytopenia.
Respiratory, thoracic and mediastinal disorders.
Churg Strauss syndrome (i.e. Eosinophilic Granulomatosis with Polyangitis).
Musculoskeletal and connective tissue disorders.
Arthralgia, myalgia, joint swelling.
Description of safety aspects of special interest.
Allergic events.
As with any protein, local or systemic allergic or Type I hypersensitivity events can occur. Frequencies of all allergic-type events were similar for both treatment groups of the total study population in adults and adolescents (4%, omalizumab, 6%, placebo). Events such as vasovagal syncope, postural hypotension, allergic bronchospasm and photosensitivity occurred in < 1% of patients in each treatment group (see Section 4.4 Special Warnings and Precautions for Use).
In post-marketing reports, the frequency of anaphylaxis in patients exposed to omalizumab use was estimated to be 0.2% based on a total number of anaphylactic reactions observed from an estimated exposure of over 500,000 patient years.
Parasitic infections.
In patients at chronic high risk of helminth infection, a placebo-controlled trial showed a numerical increase in infection rate with omalizumab that was not statistically significant. The course, severity, and response to treatment of infections were unaltered (see Section 4.4 Special Warnings and Precautions for Use, Other IgE-associated disorders).
Malignancies.
During initial clinical trials for adults and adolescents 12 years of age and older, there was a numerical imbalance in cancers arising in the active treatment group (25 patients of 5015), compared with the control group (5 patients of 2854). The number of observed cases was uncommon (< 1/100) in both the active (0.5%) and the control group, (0.2%). The observed cases in omalizumab-treated patients included breast, non-melanoma skin, prostate, melanoma and parotid malignancies. The overall observed incidence rate of malignancy in the initial omalizumab clinical trial programme was comparable to that reported in the general population (0.4%).
In a subsequent observational study comparing 5007 omalizumab-treated and 2829 non-omalizumab-treated patients followed for up to 5 years, the incidence rates of primary malignancies per 1000 patient years were 16.01 (295/18426 patient years) and 19.07 (190/9963 patient years), respectively, which does not indicate an increased malignancy risk (rate ratio 0.84, 95% confidence interval, 0.62-1.13). In a further analysis of randomised, double-blind, placebo-controlled clinical trials including 4254 patients on omalizumab and 3178 patients on placebo, omalizumab treatment was not associated with an increased malignancy risk based on incidence rates per 1000 patient years of 4.14 (14/3382 patient years) for omalizumab treated patients and 4.45 (11/2474 patient years) for placebo patients (rate ratio 0.93, 95% confidence interval 0.39-2.27).
Arterial thromboembolic events (ATE).
In controlled clinical trials and during interim analyses of an observational study, a numerical imbalance of ATEs was observed. ATE included stroke, transient ischemic attack, myocardial infarction, unstable angina, and cardiovascular death (including death from unknown cause). In the final analysis of the observational study, a numerical imbalance of ATEs was observed for: transient ischemic attack, myocardial infarction, unstable angina, and cardiovascular death (including death from unknown cause) while no notable numerical imbalance was observed for stroke. The rate of ATE per 1000 patient years was 7.52 (115/15286 patients years) for omalizumab-treated patients and 5.12 (51/9963 patient years) for control patients. In a multivariate analysis controlling for available baseline cardiovascular risk factors, the hazard ratio was 1.32 (95% confidence interval 0.91-1.91). In a new analysis of pooled clinical trials including all randomised double-blind, placebo-controlled clinical trials of 8 or more weeks duration, the rate of ATE per 1000 patient years was 2.69 (5/1856 patients years) for omalizumab-treated patients and 2.38 (4/1680 patient years) for placebo patients (rate ratio 1.13, 95% confidence interval 0.24-5.71).
Thrombocytopenia.
In clinical trials 0.6% of adult and adolescent patients experienced platelet counts below the lower limit of the normal laboratory range. None of these changes was associated with bleeding episodes or a decrease in haemoglobin. No pattern of persistent decrease in platelet counts has been reported in humans (patients greater than 6 years of age), as observed in non-human primates. (See Section 4.4 Special Warnings and Precautions for Use.)
Other laboratory data.
There was no evidence of clinically relevant changes in laboratory safety tests during clinical trials.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
No cases of overdose have been reported. A maximum tolerated dose of Omlyclo has not been determined. Single intravenous doses up to 4000 mg have been administered to patients without evidence of dose-limiting toxicities. The highest cumulative dose administered to patients was 44,000 mg over a 20-week period and this dose did not result in any untoward acute effects.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
General characteristics.
Omalizumab is a recombinant DNA-derived humanised monoclonal antibody that selectively binds to human immunoglobulin E (IgE). The antibody is an IgG1 kappa that contains human framework regions with the complementary-determining regions of a humanised murine antibody that binds to IgE.
Patients with allergic asthma and chronic rhinosinusitis with nasal polyps (CRSwNP).
IgE plays a major role in the pathophysiology of inflammatory diseases in the airways. The allergic cascade is initiated when IgE bound to the high affinity IgE receptor FcԑRI, on the surface of mast cells and basophils is crosslinked by allergen. This results in the degranulation of these effector cells and the release of histamines, leukotrienes, cytokines and other mediators. These mediators are causally linked to the pathophysiology of asthma, including airway oedema, smooth muscle contraction and altered cellular activity associated with the inflammatory process. They also contribute to the signs and symptoms of allergic asthma such as bronchoconstriction, mucous production, wheezing, dyspnoea and chest tightness.
Omalizumab binds to IgE at the same site as the high-affinity FcԑRI receptor, thereby reducing the amount of free IgE that is available to bind to the receptor. Treatment with omalizumab also reduces the number of FcԑRI receptors on basophils in atopic subjects and histamine release was reduced in response to allergen challenge in those subjects. Treatment with omalizumab inhibits IgE-mediated inflammation, as evidenced by reduced blood and tissue eosinophils and reduced inflammatory mediators, including IL-4, IL-5, and IL-13 by innate, adaptive and non-immune cells.
Patients with allergic asthma.
Clinical studies in asthma patients' showed that, serum free IgE levels (e.g. unbound IgE) are reduced in a dose dependent manner within 2 hours of subcutaneous dosing. Average decreases were 84-99% of baseline.
Serum total IgE levels (e.g. bound or unbound) increased an average of 4-fold post-dosing due to formation of omalizumab-IgE binding. Following discontinuation of omalizumab dosing, increases in total IgE and decreases in free IgE were reversible with no rebound in IgE levels after drug washout.
Patients with chronic rhinosinusitis with nasal polyps (CRSwNP).
In clinical studies in patients with CRSwNP, omalizumab treatment led to a reduction in serum free IgE and an increase in serum total IgE levels, similar to the observations in patients with allergic asthma. After repeated dosing every 2 or 4 weeks, with dosage and frequency according to Table 2 (see Section 4.2 Dose and Method of Administration), mean pre-dose serum free IgE levels decreased by approximately 95% and remained stable between 16 and 24 weeks of treatment. Total IgE levels in serum increased due to the formation of omalizumab-IgE complexes, which have a slower elimination rate compared with free IgE. After repeated dosing every 2 or 4 weeks, with dosage and frequency according to Table 2 (see Section 4.2 Dose and Method of Administration), mean pre-dose serum total IgE levels at Week 16 were 3- to 4-fold higher compared with pre-treatment levels, and remained stable between 16 and 24 weeks of treatment.
Patients with chronic spontaneous urticaria (CSU).
There are several theories for the etiology of CSU, including one that suggests an autoimmune origin. Autoimmune antibodies to IgE and its receptor, FcεRI, have been isolated from the serum of some patients with CSU. These autoantibodies can activate basophils or mast cells leading to release of histamine.
One hypothesis for the mechanism of action of omalizumab in CSU is that it lowers free IgE levels in the blood and subsequently in the skin. This leads to down-regulation of surface IgE receptors, thereby decreasing downstream signaling via the FcεRI pathway, resulting in suppressed cell activation and inflammatory responses. As a consequence, the frequency and severity of symptoms of CSU are lessened. Another hypothesis is that lowering circulating free IgE levels leads to a rapid and non-specific desensitisation of cutaneous mast cells. Down-regulation of FcεRI may help to sustain the response.
In clinical studies in CSU patients, omalizumab treatment led to a dose-dependent reduction of free IgE and an increase of total IgE levels in serum, similar to the observations in allergic asthma patients. Maximum suppression of free IgE was observed 3 days after the first subcutaneous dose. After repeated dosing once every 4 weeks, pre-dose serum free IgE levels remained stable between 12 and 24 weeks of treatment. Total IgE levels in serum increased after the first dose due to the formation of omalizumab: IgE complexes which have a slower elimination rate compared with free IgE. After repeated dosing once every 4 weeks at 75 mg to 300 mg, average pre-dose serum total IgE levels at week 12 were two-to three-fold higher compared with pre-treatment levels and remained stable between 12 and 24 weeks of treatment. After discontinuation of omalizumab, free IgE levels increased and total IgE levels decreased towards pre-treatment levels over a 16-week treatment-free follow-up period.
Clinical trials.
Allergic asthma.
Clinical trials with Xolair.
Adults and adolescents ≥ 12 years of age. The efficacy and safety of omalizumab were demonstrated in a 28-week pivotal, placebo-controlled study (study 2306) involving 419 severe allergic asthmatics, ages 12-79 years, who had reduced lung function (FEV1 40-80% predicted) and poor asthma symptom control despite receiving > 1000 micrograms of beclomethasone dipropionate (or equivalent) plus long-acting beta-2-agonist. Eligible patients had experienced multiple asthma exacerbations requiring systemic corticosteroid treatment or had been hospitalised or attended an emergency room due to a severe asthma exacerbation in the past year despite continuous treatment with high-dose inhaled corticosteroids and long-acting beta-2-agonist. Subcutaneous omalizumab or placebo were administered as add-on therapy to > 1000 micrograms inhaled beclomethasone dipropionate (or equivalent) plus long-acting beta2-agonist. Oral corticosteroid (22%), theophylline (27%) and anti-leukotriene (35%) maintenance therapies were allowed. In the treatment phase concomitant asthma therapy was not changed.
The rate of asthma exacerbations requiring treatment with systemic corticosteroids was the primary endpoint. The exacerbation rate was 0.74 on omalizumab and 0.92 on placebo and these did not differ significantly (p=0.153), however there was a difference between groups in this baseline exacerbation rate. When the analysis was adjusted for this baseline imbalance, the exacerbation rate was 0.68 on omalizumab and 0.91 on placebo (p=0.042). This approximates to a 74% (95% CIs 55%-99%) treatment effect ratio favouring omalizumab over the 28-week treatment period. Severe exacerbations (lung function less than 60% of personal best) were halved (49 omalizumab vs 100 placebo, p=0.008) resulting in 43.9% fewer asthma-related emergency visits comprised of hospitalisations, emergency room, and unscheduled doctor visits (p=0.038). The reduction in exacerbations in omalizumab-treated patients was seen in the context of statistically significant improvements in asthma symptoms, quality of life and lung function.
There were four large placebo-controlled supportive studies in adults and adolescents (> 90% meeting global criteria of severe persistent asthma) (Studies 2304, 008, 009 and 011) and one further randomised standard therapy controlled study (study IA04) which most closely matched the population in study 2306. Studies 2304, 008, 009 and IA04 used exacerbation as primary endpoint, whereas study 011 primarily evaluated inhaled corticosteroid sparing.
In study 2304 the safety and efficacy of omalizumab were demonstrated in 405 patients with co-morbid allergic asthma and perennial allergic rhinitis. Eligible patients had both symptomatic allergic asthma and perennial allergic rhinitis. Patients were treated with omalizumab or placebo for 28 weeks as add-on therapy to ≥ 400 micrograms of inhaled budesonide. Inhaled long-acting beta2-agonists (39%) and nasal corticosteroids (17%) were allowed.
The co-primary endpoints for study 2304 were the incidence of asthma exacerbations (worsening of asthma requiring systemic corticosteroids or a doubling of the patient's baseline budesonide dose) and the proportion of patients in each treatment group with a ≥ 1.0 improvement from baseline at the end of the treatment phase in both asthma and rhinitis specific quality of life assessments (Juniper Quality of Life Assessment).
Patients treated with omalizumab had a significantly lower incidence of asthma exacerbations than patients receiving placebo (20.6% omalizumab vs 30.1% placebo, p=0.02) and there was a significantly higher proportion of omalizumab-treated than placebo patients that improved by ≥ 1.0 points in both asthma and rhinitis specific quality of life assessments (57.7% omalizumab vs 40.6% placebo, p < 0.0001).
The reduction in exacerbations and improvements of quality of life in omalizumab-treated patients were seen in the context of statistically significant improvements in both rhinitis and asthma symptoms, and lung function, compared to placebo.
In two identical 16-week studies (008 and 009), the safety and efficacy of omalizumab as add-on therapy were demonstrated in 1,071 allergic asthmatics, who were symptomatic despite treatment with inhaled corticosteroids (beclomethasone dipropionate 500 to 1,200 micrograms/day).
In both trials omalizumab was superior to placebo with respect to the primary variable of asthma exacerbation (worsening of asthma requiring systemic corticosteroids or a doubling of the patient's baseline beclomethasone dose). The number of asthma exacerbations was significantly lower in the omalizumab group (p=0.006 and p < 0.001 in studies 008 and 009, respectively). Fewer omalizumab-treated patients experienced asthma exacerbations (14.6% vs 23.3%, p=0.009 in study 008 and 12.8% vs 30.5%, p < 0.001 in study 009).
In double-blind extension phases of both studies out to one year the reduction in the frequency of asthma exacerbations for omalizumab-treated patients compared to placebo-treated patients was maintained.
Study IA04 was a randomised, controlled, open-label study for 52 weeks in 312 adult and adolescent patients with poorly controlled allergic asthma. Patients received omalizumab as add-on to current asthma treatment (median dose of inhaled corticosteroids was 2000 micrograms/day, 78% were receiving a long-acting beta2-agonist) or current asthma treatment alone. Patients had to have at least one asthma-related hospitalisation or emergency room visit and at least one additional course of oral corticosteroids due to asthma in the previous year.
Treatment with omalizumab led to a 61% reduction in clinically significant asthma exacerbation rate (p < 0.001) compared to current asthma therapy alone. This reduction in exacerbations was seen in the context of statistically significant improvements in asthma symptoms, lung function and rescue medication use.
In study 011 the safety and corticosteroid-sparing effect of omalizumab was demonstrated in 246 patients with severe allergic asthma requiring daily treatment with high-dose inhaled corticosteroids (fluticasone ≥ 1000 micrograms/day) and in whom long-acting beta2-agonists were allowed. The study included a 16-week steroid stable phase with study medication added, followed by a 16-week steroid reduction phase.
The percent reduction in inhaled corticosteroid dose at the end of the treatment phase was significantly greater in omalizumab-treated patients versus placebo patients (median 60% vs. 50%, p=0.003). The proportion of omalizumab patients who were able to reduce their fluticasone dose to ≤ 500 micrograms/day was 60.3% versus 45.8% in the placebo group (p < 0.05).
The clinically meaningful treatment differences in exacerbation rates were comparable for all studies. Table 7 provides annualised exacerbation rates for each study.
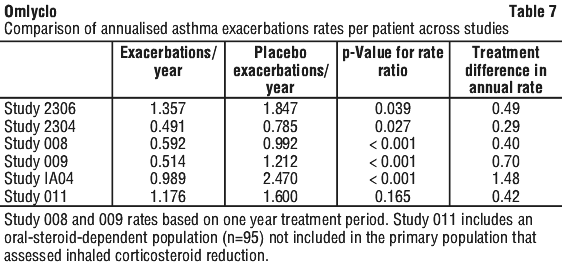 In a subgroup analysis of study 2306 (conducted in subjects with severe asthma), patients with pre-treatment total IgE < 76 IU/mL were less likely to experience a clinically meaningful benefit. In these patients omalizumab did not significantly reduce asthma exacerbation compared to placebo, whereas in patients with pre-treatment total IgE > 76IU/L, the rate of asthma exacerbations was reduced by 40% (p = 0.002).
In a subgroup analysis of study 2306 (conducted in subjects with severe asthma), patients with pre-treatment total IgE < 76 IU/mL were less likely to experience a clinically meaningful benefit. In these patients omalizumab did not significantly reduce asthma exacerbation compared to placebo, whereas in patients with pre-treatment total IgE > 76IU/L, the rate of asthma exacerbations was reduced by 40% (p = 0.002).
There are no efficacy data in patients with mild asthma (eg. those not already on inhaled steroids).
In the clinical trials of omalizumab, all subjects had a positive skin test or serum IgE RAST to a relevant aeroantigen. The efficacy of omalizumab in subjects who are negative for these tests is unknown.
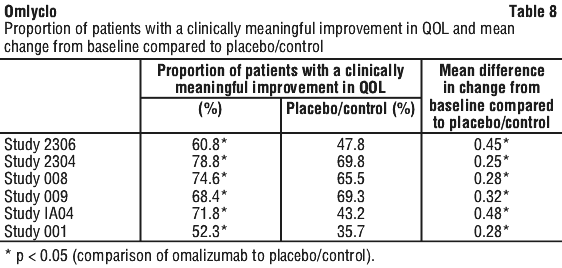
Quality of life.
Asthma-related quality of life scores were measured using the Juniper Quality of Life assessments. For all six studies there was a statistically significant improvement from baseline in Quality of Life scores for omalizumab patients versus the placebo or control group (Table 8). Improvements were demonstrated in all four asthma-specific domains of the asthma quality of life questionnaire - symptoms, activities, emotional function and environmental exposure - as well as in the overall score. In five of the six studies, a statistically significantly higher number of omalizumab patients than control patients showed a clinically meaningful improvement (≥ 0.5 points) in total Quality of Life score.
Clinical trials with Xolair.
Children 6 to < 12 years of age. The primary support for safety and efficacy of omalizumab in the 6 to < 12 years of age group comes from one randomised, double-blind, placebo controlled, multi-centre trial (study IA05).
Study IA05 was a 52 week study that evaluated the safety and efficacy of omalizumab as add-on therapy in 628 moderate to severe allergic asthmatics who were uncontrolled despite treatment with regular inhaled corticosteroids (fluticasone propionate DPI ≥ 200 microgram/day or equivalent) with or without other controller asthma medications. Eligible patients were those with a diagnosis of asthma > 1 year, a positive skin prick test to at least one perennial aeroallergen, and a history of clinical features of moderate to severe persistent asthma, including daytime and/or night-time symptoms along with a history of experiencing exacerbations within the year prior to study entry. Long-acting beta2-agonists (67.4%), anti-leukotriene (36.6%) and oral corticosteroid (1.3%) maintenance therapies were allowed. During the first 24 weeks of treatment, a patient's steroid doses remained constant from baseline and this was followed by a 28 week period during which inhaled corticosteroid adjustment was allowed.
The primary efficacy objective was to demonstrate the effect of omalizumab on the clinically significant asthma exacerbation rate during the 24 week double-blind fixed steroid treatment. A clinically significant exacerbation was defined as a worsening of asthma symptoms as judged clinically by the investigator, requiring doubling of the baseline inhaled corticosteroid dose for at least 3 days and/or treatment with rescue systemic (oral or IV) corticosteroids for at least 3 days.
Patients receiving omalizumab had a statistically significantly lower rate of asthma exacerbations than patients receiving placebo (rate ratio 0.693, p=0.007). The difference in exacerbation rates between omalizumab and placebo represents a 31% relative decrease in exacerbations. During the entire 52-week treatment period, patients receiving omalizumab had a statistically significantly lower rate of asthma exacerbations than patients receiving placebo (rate ratio 0.573, p < 0.001), which represents a 43% relative decrease in exacerbations.
After 24 weeks of treatment, reductions in mean nocturnal symptom score and rescue medication use were numerically greater in omalizumab-treated patients compared with placebo.
A pre-specified subgroup of patients with inadequately controlled severe asthma despite receiving high-dose ICS (fluticasone propionate DPI ≥ 500 microgram/day or equivalent) and a LABA, with or without other controller medications (n=235), was analysed. After 24 weeks, patients in this subgroup who received omalizumab demonstrated a statistically significantly lower rate of asthma exacerbations than patients receiving placebo (rate ratio 0.662, p=0.047), which represents a 34% relative decrease in exacerbations. In the second 28-week treatment period, patients in this subgroup who received omalizumab had a statistically significantly lower rate of asthma exacerbations than patients receiving placebo (rate ratio 0.37, p < 0.001), which represents a 63% relative decrease in exacerbations. During the entire 52-week double-blind treatment period, patients in this subgroup who received omalizumab had a statistically significantly lower rate of asthma exacerbations than patients receiving placebo (rate ratio 0.504, p < 0.001), which represents a 50% relative decrease in exacerbations.
Efficacy in patients 6 to < 12 years of age with more moderate asthma (Study 010) was apparent with reductions in exacerbation rates that were comparable to those observed in the more severe patient population (Study IA05).
Study 010 was a 28 week double blind controlled study in 334 patients who were well controlled with inhaled corticosteroids. During the first 16 weeks patients' steroid doses remained constant from baseline followed by a 12 week steroid reduction period. The rate of clinically significant asthma exacerbations during the 16-week fixed steroid treatment period was statistically significantly lower in omalizumab patients compared with placebo (rate ratio 0.577, p=0.033), representing a 42% relative decrease for omalizumab patients. The rate of clinically significant asthma exacerbations during the 28-week double-blind treatment period was statistically significantly lower in omalizumab patients compared with placebo (p < 0.001), representing a relative decrease of 50%.
The percentage reduction of ICS dose at the end of the 28-week treatment period was statistically significantly greater for omalizumab patients (p=0.013). At the end of the 28-week treatment period, greater benefits were observed in change from baseline Paediatric Asthma Quality of Life Questionnaire (PAQLQ) for omalizumab-treated patients compared to placebo-treated patients (p=0.030).
Clinical trials with Xolair.
Chronic rhinosinusitis with nasal polyps (CRSwNP). The safety and efficacy of omalizumab were evaluated in two randomized, multicenter, double-blind, placebo-controlled clinical trials that enrolled patients with CRSwNP (study 1, N=138; study 2, N=127). Patients received omalizumab or placebo subcutaneously every 2 or 4 weeks, with dosage and frequency according to Table 2 (see Section 4.2 Dose and Method of Administration). All patients received background intranasal mometasone therapy throughout the study. Prior sino-nasal surgery or prior systemic corticosteroid usage were not required for inclusion in the studies. Patients received omalizumab or placebo for 24 weeks followed by a 4-week follow-up period. Demographics and baseline characteristics, including allergic comorbidities, are described in Table 9.
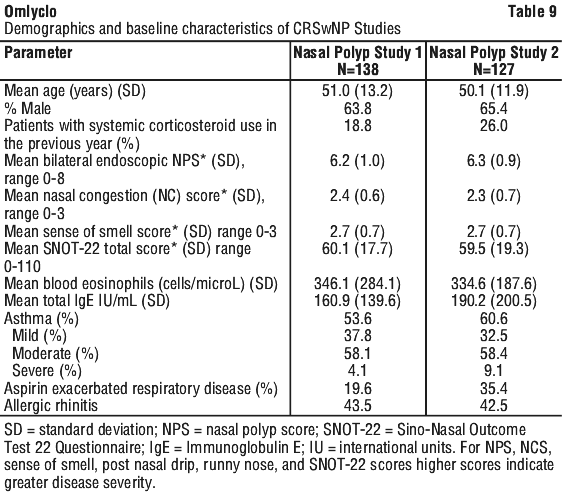 The co-primary endpoints were bilateral nasal polyp score (NPS) and average daily nasal congestion score (NCS) at Week 24. NPS was measured via endoscopy at baseline and pre-specified time points and scored (range 0-4 per nostril) for a total NPS (range 0/best-8/worst). Nasal congestion was measured by a daily NCS (range 0/best-3/worst). Patients were required to have NPS ≥ 5 and weekly average of NCS > 1 prior to randomization, despite use of intranasal mometasone. The mean NPS at baseline was balanced between the two treatment groups in both studies.
The co-primary endpoints were bilateral nasal polyp score (NPS) and average daily nasal congestion score (NCS) at Week 24. NPS was measured via endoscopy at baseline and pre-specified time points and scored (range 0-4 per nostril) for a total NPS (range 0/best-8/worst). Nasal congestion was measured by a daily NCS (range 0/best-3/worst). Patients were required to have NPS ≥ 5 and weekly average of NCS > 1 prior to randomization, despite use of intranasal mometasone. The mean NPS at baseline was balanced between the two treatment groups in both studies.
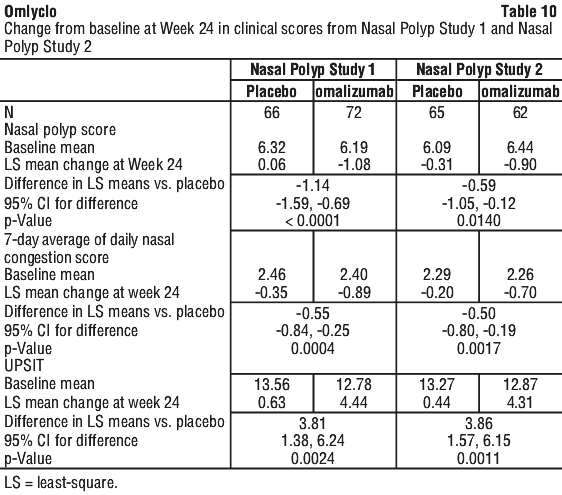
In both CRSwNP studies 1 and 2, patients who received omalizumab had a statistically significant greater improvement from baseline at Week 24 in NPS and weekly average NCS than patients who received placebo. Results from Nasal Polyps Study 1 and 2 are shown in Table 10.
The greater improvements in NPS and NCS in the omalizumab group compared to the placebo group were observed as early as the first assessment at Week 4 in both studies as seen in Figure 1. The LS mean difference in change from baseline at Week 4 in NPS in omalizumab compared to placebo was -0.92 (95% CI:-1.37, -0.48) in study 1 and -0.52 (95% CI: -0.94, -0.11) in study 2. The LS mean difference in change from baseline at Week 4 in NCS in omalizumab compared to placebo was -0.25 (95% CI: -0.46, -0.04) in study 1 and -0.26 (95% CI: -0.45, -0.07) in study 2. However, statistical tests at this time point were not pre-specified.
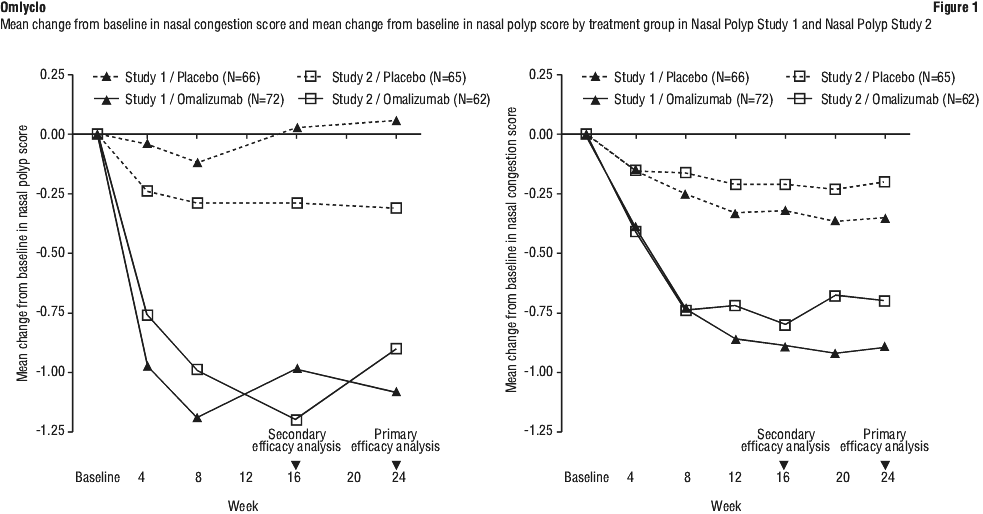 A key secondary endpoint was the assessment of the change from baseline at Week 24 of the total nasal symptom score (TNSS). Patient-reported TNSS was the sum of four equally weighted individual daily symptom scores: NCS, sense of smell score, posterior rhinorrhea score, and anterior rhinorrhea score. The TNSS ranged from 0/best-12/worst. omalizumab significantly improved the average daily TNSS compared to placebo. The LS mean difference for change from baseline to Week 24 was -1.91 points (95% CI: -2.85, -0.96; p = 0.0001) in study 1 and -2.09 points (95% CI: -3.00, -1.18; p < 0.0001) in study 2.
A key secondary endpoint was the assessment of the change from baseline at Week 24 of the total nasal symptom score (TNSS). Patient-reported TNSS was the sum of four equally weighted individual daily symptom scores: NCS, sense of smell score, posterior rhinorrhea score, and anterior rhinorrhea score. The TNSS ranged from 0/best-12/worst. omalizumab significantly improved the average daily TNSS compared to placebo. The LS mean difference for change from baseline to Week 24 was -1.91 points (95% CI: -2.85, -0.96; p = 0.0001) in study 1 and -2.09 points (95% CI: -3.00, -1.18; p < 0.0001) in study 2.
The SNOT-22 (Sino-Nasal Outcome Test), combines measures of sinonasal symptoms with psychological and sleep dysfunction measures and has a score that ranges from 0 to 110 (0/best-110/worst). The minimal clinical important difference has been established as 8.9. The proportion of patients who experienced an improvement from baseline at Week 24 of at least the MID (8.9 points) in SNOT-22 score for the individual studies was: Study GA39688 - 30/65 (46.2%) in the placebo arm and 53/69 (76.8%) in the omalizumab arm, OR of 4.55 (95% CI 2.07, 9.97; p=0.0002); Study GA39855in and 23/63 (36.5%) in the placebo arm and 39/59 (66.1%) in the omalizumab arm, OR 3.71 (95% CI: 1.72, 8.04; p=0.0009).
Additional secondary endpoint analyses included Week 16 assessments of NPS and NCS. Omalizumab significantly improved the NPS at week 16, (range 0/best-8/worst) compared to placebo. The LS mean difference for change from baseline to Week 16 in omalizumab compared to placebo was -1.01 (95% CI: - 1.43, -0.60; p < 0.0001) in study 1 and -0.91 (95% CI: -1.39, -0.44; p = 0.0002) in study 2. Omalizumab significantly improved the NCS at week 16, (range 0/best-3/worst) compared to placebo. The LS mean difference for change from baseline to Week 16 in average daily NCS in omalizumab compared to placebo was -0.57 (95% CI: -0.83, -0.31; p < 0.0001) in study 1 and of -0.59 (95% CI: -0.87, -0.30; p < 0.0001) in study 2.
In a pre-specified pooled analysis of rescue treatment (systemic corticosteroids for ≥ 3 consecutive days or nasal polypectomy) during the 24-week treatment period, the proportion of patients requiring rescue treatment was lower in omalizumab compared to placebo (2.3% versus 6.2%, respectively). The treatment arm difference was not statistically significant (OR: 0.38; 95% CI: 0.10, 1.49; nominal p=0.1639). There were no sino-nasal surgeries reported in either study.
Clinical trials with Xolair.
Chronic spontaneous urticaria (CSU). The efficacy and safety of omalizumab were demonstrated in two randomised, placebo-controlled phase III studies (study Q4881g, Q4882g) in patients with CSU who remained symptomatic despite H1 antihistamine therapy at the approved dose. All patients received omalizumab or placebo in addition to H1 antihistamines. A third study (Q4883g) primarily evaluated the safety of omalizumab in patients with CSU who remained symptomatic despite treatment with H1 antihistamines at increased doses and H2 antihistamine and/or leukotriene receptor antagonist (LTRA) treatment. All patients received omalizumab or placebo in addition to H1 antihistamines up to 4 times the approved dose, and/or H2 antihistamines and/or LTRAs. CSU patients with external triggers were excluded from these trials. The three studies enrolled 975 patients aged between 12 and 75 years (mean age 42.3 years; 39 patients 12-17 years, 54 patients ≥ 65 years; 259 males and 716 females). All patients were required to have inadequate symptom control, as assessed by a weekly urticaria activity score (UAS7, range 0 42) of ≥ 16, and a weekly itch severity score (which is a component of the UAS7; range 0 21) of ≥ 8 for the 7 days prior to randomisation, despite having used an antihistamine for at least 2 weeks beforehand.
In studies Q4481g and Q4882g, patients had a mean weekly itch severity score of between 13.7 and 14.5 at baseline and a mean UAS7 score of 29.5 and 31.7 respectively. Patients in safety study Q4883g had a mean weekly itch severity score of 13.8 and a mean UAS7 score of 31.2 at baseline. Across all three studies, patients reported receiving on average 4 to 6 medications (including H1 antihistamines) for CSU symptoms prior to study enrolment. Patients received omalizumab at 75 mg, 150 mg or 300 mg or placebo by subcutaneous injection every 4 weeks for 24 and 12 weeks in studies 1 and 2, respectively, and 300 mg or placebo by subcutaneous injection every 4 weeks for 24 weeks in study 3. All studies had a 16 week treatment-free follow-up period. See Table 11.
 In studies Q4881g and Q4882g the 75 mg dose did not consistently meet either the primary efficacy endpoint or a number of secondary endpoints. It was deemed not efficacious and therefore not further presented.
In studies Q4881g and Q4882g the 75 mg dose did not consistently meet either the primary efficacy endpoint or a number of secondary endpoints. It was deemed not efficacious and therefore not further presented.
Change from baseline to week 12 in weekly itch severity score.
The primary efficacy endpoint, change from baseline to week 12 in weekly itch severity score was met by both the 150 mg and 300 mg doses in studies Q4881g and Q4882g and by the 300 mg dose in Q4883g (see Table 12).
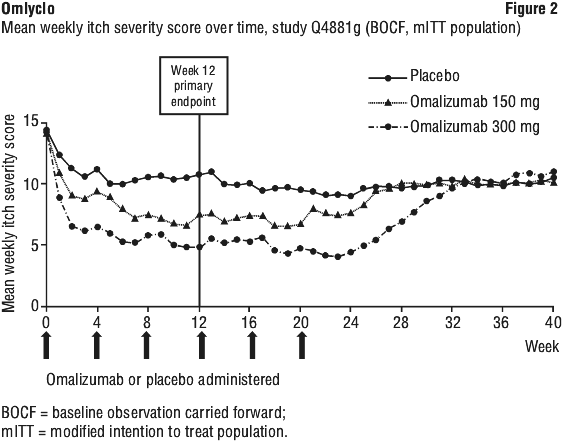
 Figure 2 shows the mean weekly itch severity score over time in study Q4881g. The mean weekly itch severity scores significantly decreased in both treatment groups with a maximum effect around week 12 that was sustained over the 24-week treatment period. In studies Q4883g (300 mg over the 24-week treatment period) and Q4882g (150 mg and 300 mg over the 12-week treatment period) the results were similar to those of study Q4881g.
Figure 2 shows the mean weekly itch severity score over time in study Q4881g. The mean weekly itch severity scores significantly decreased in both treatment groups with a maximum effect around week 12 that was sustained over the 24-week treatment period. In studies Q4883g (300 mg over the 24-week treatment period) and Q4882g (150 mg and 300 mg over the 12-week treatment period) the results were similar to those of study Q4881g.
In all three studies (see Figure 2 for study Q4881g), the mean weekly itch severity score for both doses increased gradually during the 16-week treatment-free follow-up period, consistent with symptom re-occurrence. Mean values at the end of the follow-up period were similar to the placebo group, but lower than respective mean baseline values.
 Secondary endpoints for studies Q4881g, Q4882g and Q4883g are presented in Table 13.
Secondary endpoints for studies Q4881g, Q4882g and Q4883g are presented in Table 13.
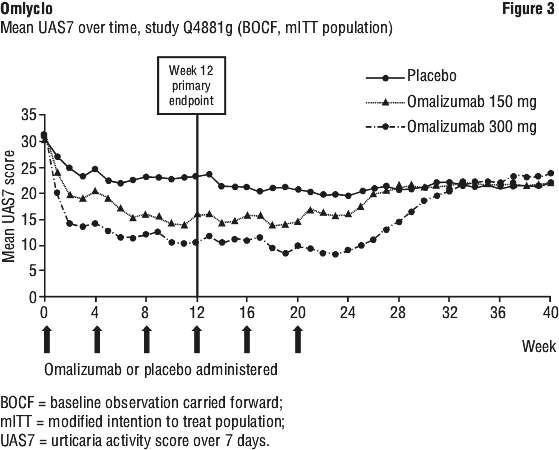
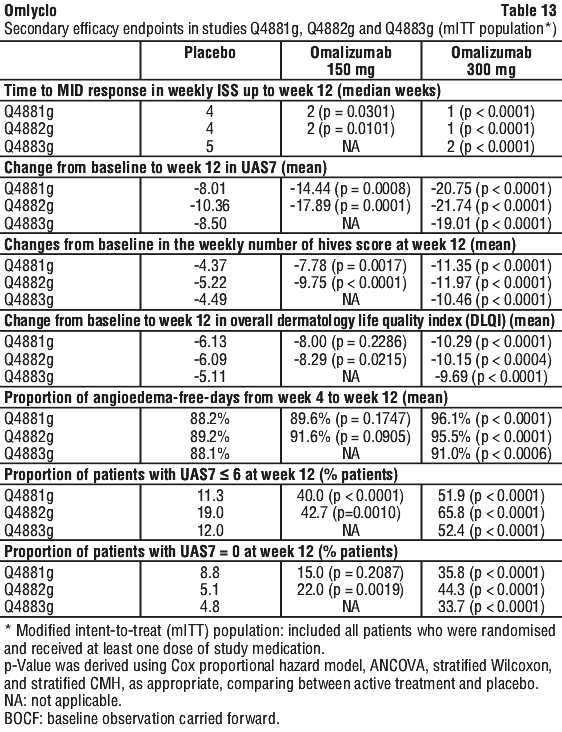 Figure 3 shows mean UAS7 over time in study Q4881g, displaying a significant decrease from baseline in both treatment groups with a maximum effect around week 12. The magnitude of the effect was maintained during the 24-week treatment period. In studies Q4882g (150 mg and 300 mg over the 12-week treatment period) and Q4883g (300 mg over 24-week treatment period) the results were similar to those of study Q4881g.
Figure 3 shows mean UAS7 over time in study Q4881g, displaying a significant decrease from baseline in both treatment groups with a maximum effect around week 12. The magnitude of the effect was maintained during the 24-week treatment period. In studies Q4882g (150 mg and 300 mg over the 12-week treatment period) and Q4883g (300 mg over 24-week treatment period) the results were similar to those of study Q4881g.
In all three studies (see Figure 3 for study Q4881g), the UAS7 for both omalizumab treatment groups increased gradually during the 16-week treatment-free follow-up period, consistent with symptom re occurrence. Mean values at the end of the follow-up period were similar to the placebo group but lower than respective mean baseline values.
Efficacy after 24 weeks of treatment.
Table 14 shows the results after 24 weeks of treatment. Similar magnitudes of response are seen as at 12 weeks.
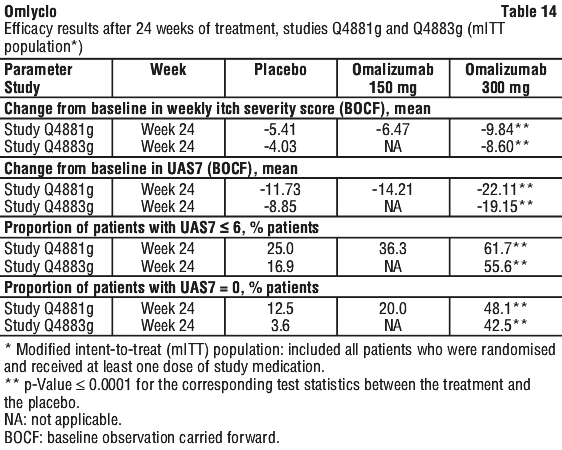 The treatment duration in the studies were 12 weeks (Q4882g) and 24 weeks, respectively (Q4881g and Q4883g), therefore the Clinical trial experience of long-term treatment beyond 6 months in this indication is limited.
The treatment duration in the studies were 12 weeks (Q4882g) and 24 weeks, respectively (Q4881g and Q4883g), therefore the Clinical trial experience of long-term treatment beyond 6 months in this indication is limited.
5.2 Pharmacokinetic Properties
General characteristics.
Absorption.
Administration of omalizumab manufactured as a lyophilised or liquid formulation resulted in similar serum concentration-time profiles of omalizumab.
The lyophilised formulation is not available in this brand but it is available in other brands.
Distribution.
In vitro, omalizumab forms complexes of limited size with IgE. The composition and molecular weight of the complexes are dependent on the molar ratio of omalizumab to IgE. Precipitating complexes and complexes larger than 1 million molecular weight were not observed in vitro. Complexes formed in vitro were similar to those studied in vivo. Tissue distribution studies in cynomolgus monkeys showed no specific uptake of 125I-omalizumab by any organ or tissue.
Metabolism.
No circulating metabolites were detected after intravenous administration of 125I-omalizumab to cynomolgus monkeys.
Excretion.
Since omalizumab is a recombinant humanised IgG1, its mechanism of clearance from the serum involves IgG clearance processes as well as clearance via specific binding and complex formation with its target ligand, free serum IgE. In studies in mice and monkeys, the omalizumab: IgE complexes were eliminated by interactions with Fcγ receptors within the liver and the reticuloendothelial system, at rates which were generally faster than IgG clearance.
Patients with allergic asthma and seasonal allergic rhinitis patients.
Absorption.
Following single, subcutaneous bolus administration, omalizumab is absorbed slowly, reaching mean peak serum concentrations after 6 to 10 days. Although not precisely defined, the mean absolute bioavailability after subcutaneous administration in humans is estimated to be approximately 53 - 71%.
Distribution.
Distribution volumes were 110 ± 14 mL/kg and typical of distribution volumes seen with large macromolecules.
Elimination.
Omalizumab has a long serum half-life (mean 22 + 8.2 days). The long half-life is characteristic of IgG class immunoglobulins and a result of IgG recycling via its salvage receptor (FcRn). At the doses recommended for therapeutic use, average clearance is expected to represent dominantly IgG clearance and to be relatively slow (2.27-4.12 mL/kg/day).
Age, gender, race.
There are no clinically important differences in pharmacokinetic and pharmacodynamic data within the 6-75 age range or by gender or race.
Patients with chronic rhinosinusitis with nasal polyps (CRSwNP).
The population pharmacokinetics analyses of omalizumab suggested that pharmacokinetics of omalizumab in CRSwNP were consistent with that in asthma. Graphical covariate analyses were performed to evaluate the effects of demographic characteristics and other factors on omalizumab exposure and clinical responses. These analyses demonstrate that no dose adjustments are necessary for age (18 to 75 years) or gender. Race and ethnicity data are too limited in CRSwNP to inform dose adjustment.
Patients with chronic spontaneous urticaria (CSU).
Absorption.
Following a single subcutaneous dose in adult and adolescent patients with CSU, omalizumab was absorbed slowly, reaching peak serum concentrations after an average of 6 to 8 days.
In patients with CSU, omalizumab exhibited linear pharmacokinetics across the dose range of 75 mg to 600 mg given as a single subcutaneous dose. Following doses of 75 mg, 150 mg or 300 mg every 4 weeks, trough serum concentrations of omalizumab increased proportionally with the dose level.
Distribution.
Based on population pharmacokinetic, distribution of omalizumab in CSU patients was similar to that in patients with allergic asthma.
Elimination.
In patients with CSU, based on population pharmacokinetic simulations, omalizumab serum elimination half-life at steady state averaged 24 days and apparent clearance at steady state averaged 240 mL/day (corresponding to 3.0 mL/kg/day for an 80 kg patient).
Age, race/ethnicity, gender, body weight, body mass index, baseline IgE, anti-FcεRI autoantibodies, co-medications.
The effects of demographic covariates and other factors on omalizumab exposure were evaluated using population pharmacokinetics. In addition, covariate effects were evaluated by analysing the relationship between omalizumab concentrations and clinical responses. These analyses suggest that no dose adjustments are necessary in patients with CSU for age (12 to 75 years), race/ethnicity, gender, body weight, body mass index, baseline IgE, Anti-FcεRI autoantibodies or concomitant use of H2 antihistamines or leukotriene receptor antagonists (LTRAs).
Patients with renal and hepatic impairment.
There are no pharmacokinetic or pharmacodynamic data in patients with renal or hepatic impairment (see Section 4.4 Special Warnings and Precautions for Use).
5.3 Preclinical Safety Data
Genotoxicity.
There was no evidence of gene mutations in a bacterial gene mutation assay with omalizumab. The clastogenic potential of omalizumab has not been investigated.
Carcinogenicity.
No long-term studies have been performed in animals to evaluate the carcinogenic potential of omalizumab.6 Pharmaceutical Particulars
6.1 List of Excipients
Solution for injection in pre-filled syringe and pre-filled pen.
Arginine hydrochloride, histidine hydrochloride monohydrate, histidine, polysorbate 20, water for injections.
6.2 Incompatibilities
Solution for injection in pre-filled syringe and pre-filled pen.
This medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Solution for injection in pre-filled syringe and pre-filled pen.
Store at 2°C to 8°C. Do not freeze. Store in the original package and protect from light. Allow the syringe or pen to reach room temperature (25°C) before injection. If necessary, the product may be returned to the refrigerator for later use. The total time that the syringe or pen is kept at room temperature (25°C) must not exceed 7 days.
Product is for single use in one patient only. Discard any residue.
6.5 Nature and Contents of Container
Solution for injection in pre-filled syringe with needle guard.
Pre-filled syringe comprising a type I glass syringe barrel with staked needle (stainless steel), rubber plunger stopper, and rigid needle shield composed of a rubber needle shield covered by a rigid shell. Each pre-filled syringe is mounted with a plastic safety device (needle guard) to prevent from needle stick injury.
Each pre-filled syringe of 0.5 mL contains 75 mg of omalizumab.
Each pre-filled syringe of 1 mL contains 150 mg of omalizumab.
Available as a single packaged pre-filled syringe and in multipacks containing either 6 or 10 individually packaged pre-filled syringes.
Not all pack sizes and presentations may be marketed.
Solution for injection in pre-filled pen.
Pre-filled pen comprising a type I glass syringe barrel with 27-gauge staked needle (stainless steel), (type I) plunger stopper (elastomer) and needle cap (elastomer and polypropylene).
Each pre-filled pen of 0.5 mL contains 75 mg of omalizumab.
Each pre-filled pen of 1 mL contains 150 mg of omalizumab.
Available as a single packaged pre-filled pen and in multipacks containing either 4, 6 or 10 individually packaged pre-filled pens.
Not all pack sizes and presentations may be marketed.
6.6 Special Precautions for Disposal
Dispose the used syringe immediately in a sharps container (closable, puncture resistant container). For the safety and health of you and others, needles and used syringes must never be re-used.
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
Not applicable.
CAS number.
0242138-07-4.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only.
Summary Table of Changes
