The Australian Commission on Safety and Quality in Health Care (the Commission) has completed its review of the NPS MedicineWise website and associated digital assets. Several key programs have already transitioned to the Commission including QUM Learning, MedicineInsight, Practice Reflections and the National Medicines Symposium. In addition, the Commission will soon be launching a new website that will include Medicine Finder and select QUM resources. All remaining resources on the NPS website will be archived and the website will be decommissioned in early May 2026. For more information, please email medsafety@safetyandquality.gov.au.
Please be aware that some content on this website may be outdated and not reflect current evidence or guidance.
Omtralo is used to treat a type of heart failure in adults. Omtralo has been shown to lower the risk of cardiovascular related deaths and heart failure related hospitalisation.
Omtralo contains the active ingredients sacubitril and valsartan. It is an angiotensin receptor neprilysin inhibitor(ARNI), which contains two active ingredients: sacubitril (aneprilysin inhibitor), and valsartan (an angiotensinreceptor blocker or ARB). Omtralo works by blocking thechanges caused by the enzyme neprilysin, and angiotensinII. As a result, blood vessels relax and less water is kept bythe body which is useful to treat heart failure.
Published by MIMS August 2025
BRAND INFORMATION
Brand name
Omtralo
Active ingredient
Sacubitril; Valsartan
Schedule
S4
Omtralo 24/26 (24.3 mg/25.7 mg) Tablets
Omtralo 49/51 (48.6 mg/51.4 mg) Tablets
Omtralo 97/103 (97.2 mg/102.8 mg) Tablets
1 Name of Medicine
Sacubitril and valsartan.
2 Qualitative and Quantitative Composition
Omtralo film-coated tablets are available in 3 strengths:
Each Omtralo 24/26 film-coated tablet contains 24.3 mg sacubitril and 25.7 mg valsartan (where both drug substances are combined as a sodium salt hydrate complex). This has been rounded to 24 mg/26 mg throughout the document.
Each Omtralo 49/51 film-coated tablet contains 48.6 mg sacubitril and 51.4 mg valsartan (where both drug substances are combined as a sodium salt hydrate complex). This has been rounded to 49 mg/51 mg throughout the document.
Each Omtralo 97/103 film-coated tablet contains 97.2 mg sacubitril and 102.8 mg valsartan (where both drug substances are combined as a sodium salt hydrate complex). This has been rounded to 97 mg/103 mg throughout the document.
For the full list of excipients, see Section 6.1 List of Excipients.
Light pink, ovaloid, biconvex, film-coated tablet with beveled edges; unscored, debossed with "NVR" on one side and "L11" on the other side.
4 Clinical Particulars
4.1 Therapeutic Indications
Omtralo is indicated in adult patients for the treatment of chronic heart failure (NYHA class II-IV) with reduced ejection fraction.
4.2 Dose and Method of Administration
Omtralo is administered in place of an ACE inhibitor or other ARB.
Omtralo should be initiated, and up-titration conducted, by a physician experienced with the treatment of heart failure.
Dosage.
The recommended starting dose of Omtralo is one tablet of 49 mg/51 mg twice daily, except in the situations described below.
The dose of Omtralo should be doubled after 2 to 4 weeks to the target maintenance dose of one tablet of 97 mg/103 mg twice daily, as tolerated by the patient.
If patients experience tolerability issues (systolic blood pressure ≤ 95 mmHg, symptomatic hypotension, hyperkalaemia, renal dysfunction), consideration should be given to adjustment of concomitant medications, or to temporary down-titration or discontinuation of Omtralo.
Starting dose of Omtralo of 24 mg/26 mg for some populations.
A starting dose of Omtralo of one tablet of 24 mg/26 mg taken twice daily is recommended for patients not currently taking an ACE inhibitor or an ARB, or patients previously taking low doses of these agents (see Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 4.2 Dose and Method of Administration, Other important considerations for dosing).
A starting dose of Omtralo of one tablet of 24 mg/26 mg taken twice daily should be considered for patients who have risk factors for hypotension, including patients ≥ 75 years old and patients with low systolic blood pressure (SBP ≥ 100 to 110 mmHg) (see Section 4.4 Special Warnings and Precautions for Use, Hypotension).
The dose of Omtralo should be doubled every 2-4 weeks to the target dose of one tablet of Omtralo 97 mg/103 mg twice daily, as tolerated by the patient.
See Special populations section below for further starting dose recommendations in Renal insufficiency, Hepatic insufficiency and geriatric patients.
Other important considerations for dosing.
Omtralo is contraindicated with concomitant use of an angiotensin-converting enzyme (ACE) inhibitor. Due to the potential risk of angioedema when used concomitantly with an ACE inhibitor, Omtralo must not be administered until 36 hours after the last dose of ACE inhibitor therapy and similarly, at least 36 hours must elapse after the last dose of Omtralo before ACE inhibitor therapy is initiated (see Section 4.3 Contraindications).
Omtralo should not be co-administered with an ARB due to the angiotensin II receptor blocking activity of Omtralo (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Treatment should not be initiated in patients with serum potassium level > 5.4 mmol/L or with SBP < 100 mmHg (see Section 4.4 Special Warnings and Precautions for Use).
The valsartan contained within Omtralo is more bioavailable than the valsartan in other marketed tablet formulations (see Table 1).
Special populations.
Renal insufficiency.
No dose adjustment is required in patients with mild (eGFR 60-90 mL/min/1.73 m2) to moderate (eGFR 30-60 mL/min/1.73 m2) renal impairment.
A starting dose of Omtralo 24 mg/26 mg twice daily is recommended in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2). Caution is recommended when using Omtralo in these patients as there are no adequate data (see Section 5.2 Pharmacokinetic Properties, Special populations).
There is no experience in patients with end-stage renal disease and use of Omtralo is not recommended.
Hepatic insufficiency.
No dose adjustment is required when administering Omtralo to patients with mild hepatic impairment (Child-Pugh A classification).
A starting dose of Omtralo 24 mg/26 mg twice daily is recommended for patients with moderate hepatic impairment (Child-Pugh B classification).
Patients with severe hepatic impairment, biliary cirrhosis or cholestasis (Child-Pugh C classification) should not take Omtralo (see Section 4.3 Contraindications; Section 5.2 Pharmacokinetic Properties, Special populations).
Method of administration.
For oral use. Omtralo may be administered with or without food (see Section 5.2 Pharmacokinetic Properties, Absorption).
4.3 Contraindications
Hypersensitivity to the active substance, sacubitril, valsartan, or to any of the excipients.
Concomitant use with ACE inhibitors. Do not administer Omtralo within 36 hours of switching from or to an ACE inhibitor (see Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Known history of angioedema related to previous ACE inhibitor or ARB therapy.
Hereditary or idiopathic angioedema (see Section 4.4 Special Warnings and Precautions for Use).
Concomitant use with aliskiren in patients with Type 2 diabetes (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Severe hepatic impairment, biliary cirrhosis and cholestasis (see Section 4.2 Dose and Method of Administration).
Pregnancy (see Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy).
4.4 Special Warnings and Precautions for Use
Dual blockade of the renin-angiotensin-aldosterone system (RAAS).
Omtralo must not be administered with an ACE inhibitor due to the risk of angioedema. Omtralo must not be initiated until 36 hours after taking the last dose of ACE inhibitor therapy. If treatment with Omtralo is stopped, ACE inhibitor therapy must not be initiated until 36 hours after the last dose of Omtralo (see Section 4.3 Contraindications; Section 4.2 Dose and Method of Administration; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Caution is required while co-administering Omtralo with direct renin inhibitors such as aliskiren (see Section 4.3 Contraindications; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Omtralo must not be administered with aliskiren in patients with Type 2 diabetes (see Section 4.3 Contraindications).
Omtralo should not be co-administered with an ARB due to the angiotensin II receptor blocking activity of Omtralo (see Section 4.2 Dose and Method of Administration; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Hypotension.
Omtralo lowers blood pressure and may cause symptomatic hypotension, especially in patients ≥ 75 years old, patients with renal disease and patients with low systolic blood pressure (< 112 mmHg) (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients with systolic blood pressure < 100 mmHg at the time of initiation of Omtralo have not been studied; use of Omtralo in these patients is not recommended. In the double-blind period of PARADIGM-HF, 18% of patients treated with Omtralo and 12% of patients treated with enalapril reported hypotension as an adverse event, with hypotension reported as a serious adverse event in approximately 1.5% of patients in both treatment arms.
When initiating therapy or during dose titration with Omtralo, blood pressure should be monitored routinely. Patients with an activated renin-angiotensin system, such as volume- and/or salt-depleted patients (e.g. those being treated with high doses of diuretics), are at greater risk.
If hypotension occurs, dose adjustment of diuretics, concomitant antihypertensive drugs, and treatment of other causes of hypotension (e.g. hypovolaemia) should be considered. If hypotension persists despite such measures, the dosage of Omtralo should be reduced or the product should be temporarily discontinued (see Section 4.2 Dose and Method of Administration). Permanent discontinuation of therapy is usually not required. Symptomatic hypotension is more likely to occur if the patient has been volume-depleted, e.g. by diuretic therapy, dietary salt restriction, diarrhea or vomiting. Sodium and/or volume depletion should be corrected before starting treatment with Omtralo.
Hyperkalaemia.
Treatment should not be initiated if the serum potassium level is > 5.4 mmol/L. Through its action on the renin‐angiotensin‐aldosterone system, hyperkalaemia may occur with Omtralo. In the double-blind period of PARADIGM-HF, 12% of patients treated with Omtralo and 14% of patients treated with enalapril reported hyperkalaemia as an adverse event (see Section 4.8 Adverse Effects (Undesirable Effects)). The incidence of clinically relevant hyperkalaemia was low, resulting in treatment discontinuation in 0.26% of Omtralo treated patients compared to 0.35% of enalapril treated patients. Monitor serum potassium periodically and treat appropriately, especially in patients with risk factors for hyperkalaemia such as severe renal impairment, diabetes, hypoaldosteronism, or a high potassium diet. Dosage reduction or interruption of Omtralo may be required (see Section 4.2 Dose and Method of Administration). Medications known to raise potassium levels (e.g. potassium-sparing diuretics, potassium supplements) should be used with caution when co-administered with Omtralo. If clinically significant hyperkalaemia occurs, measures such as reducing dietary potassium, or adjusting the dose of concomitant medications should be considered. In addition, if serum potassium level is > 5.4 mmol/L, discontinuation of Omtralo should be considered.
Angioedema.
Angioedema has been reported in 0.5% of patients treated with Omtralo and 0.2% of patients treated with enalapril in PARADIGM-HF. If angioedema occurs, Omtralo should be immediately discontinued and appropriate therapy and monitoring should be provided until complete and sustained resolution of signs and symptoms has occurred. Omtralo must not be re-administered. In cases of confirmed angioedema where swelling has been confined to the face and lips, the condition has generally resolved without treatment, although antihistamines have been useful in relieving symptoms.
Angioedema associated with laryngeal edema may be fatal. Where there is involvement of the tongue, glottis or larynx, likely to cause airway obstruction, appropriate therapy, e.g. subcutaneous epinephrine/adrenaline solution 1:1000 (0.3 mL to 0.5 mL) and/or measures necessary to ensure a patent airway, should be promptly administered.
Patients with a prior history of angioedema were not studied. As they may be at higher risk for angioedema, caution is recommended if Omtralo is used in these patients. Omtralo must not be used in patients with a known history of angioedema related to previous ACE inhibitor or ARB therapy or with hereditary or idiopathic angioedema (see Section 4.3 Contraindications).
Black patients may have increased susceptibility to develop angioedema.
Patients with renal artery stenosis.
Similar to other drugs that affect the renin-angiotensin-aldosterone system, Omtralo may increase blood urea and serum creatinine levels in patients with bilateral or unilateral renal artery stenosis. Caution is required in patients with renal artery stenosis and monitoring of renal function is recommended.
Patients with NYHA functional classification IV.
Caution should be exercised when initiating Omtralo in patients with NYHA functional classification IV due to limited clinical experience in this population.
Use in hepatic impairment.
There is limited clinical experience in patients with moderate hepatic impairment (Child-Pugh B classification) or with AST/ALT values more than twice the upper limit of the normal range. In these patients, exposure may be increased and safety is not established. Caution is therefore recommended when using it in these patients (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties). Omtralo is contraindicated in patients with severe hepatic impairment, biliary cirrhosis or cholestasis (Child-Pugh C classification) (see Section 4.3 Contraindications).
Use in renal impairment.
As a consequence of inhibiting the renin-angiotensin-aldosterone system, the use of Omtralo may be associated with decreased renal function. In the double-blind period of PARADIGM-HF, 5% of patients in both the Omtralo and enalapril groups reported renal failure as an adverse event (see Section 4.8 Adverse Effects (Undesirable Effects)). The incidence of clinically relevant renal impairment was low and associated treatment discontinuation was observed less frequently in patients receiving Omtralo (0.65%) compared to enalapril (1.28%). In patients whose renal function depends upon the activity of the renin-angiotensin-aldosterone system (e.g. patients with severe congestive heart failure), treatment with ACE inhibitors and angiotensin receptor antagonists has been associated with oliguria, progressive azotemia and, rarely, acute renal failure and death.
Use of Omtralo should include appropriate assessment of renal function, before initiation of therapy, and then during treatment, as appropriate. Closely monitor serum creatinine, and down-titrate or interrupt Omtralo in patients who develop a clinically significant decrease in renal function (see Section 5.2 Pharmacokinetic Properties, Special populations). As with all drugs that affect the RAAS, Omtralo may increase blood urea and serum creatinine levels in patients with bilateral or unilateral renal artery stenosis. In patients with renal artery stenosis, monitor renal function.
Patients with mild and moderate renal impairment are more at risk of developing hypotension. There is very limited clinical experience in patients with severe renal impairment (estimated GFR < 30 mL/min/1.73 m2) and these patients may be at greatest risk of hypotension. Caution should be exercised when administering Omtralo in patients with severe renal impairment. There is no experience in patients with end-stage renal disease and use of Omtralo is not recommended (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties, Special populations).
Use in the elderly.
No dose adjustment is required in patients over 65 years. However, Omtralo has been studied in a limited number of patients over 80 years. In patients ≥ 75 years old, a starting dose of one tablet of Omtralo 24 mg/26 mg taken twice daily should be considered.
Paediatric use.
The safety and efficacy of Omtralo in paediatric patients aged below 18 years have not been established.
Effects on laboratory tests.
No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Anticipated interactions resulting in a contraindication.
ACE inhibitors.
The concomitant use of Omtralo with ACE inhibitors is contraindicated, as the concomitant inhibition of neprilysin (NEP) and ACE inhibitor therapy may increase the risk of angioedema. Omtralo must not be started until 36 hours after taking the last dose of ACE inhibitor therapy. ACE inhibitor therapy must not be started until 36 hours after the last dose of Omtralo (see Section 4.3 Contraindications; Section 4.2 Dose and Method of Administration).
Aliskiren.
The concomitant use of Omtralo with aliskiren is contraindicated in patients with Type 2 diabetes (see Section 4.3 Contraindications). Combination of Omtralo with aliskiren is potentially associated with a higher frequency of adverse events such as hypotension, hyperkalaemia and decreased renal function (including acute renal failure).
Anticipated interactions resulting in concomitant use not being recommended.
Omtralo should not be co-administered with an ARB due to the angiotensin II receptor blocking activity of Omtralo (see Section 4.4 Special Warnings and Precautions for Use).
Concomitant use with aliskiren should be avoided in patients with renal impairment (eGFR < 60 mL/min/1.73 m2) (see Section 4.4 Special Warnings and Precautions for Use).
Observed interactions to be considered.
Statins.
In vitro data indicates that sacubitril inhibits OATP1B1 and OATP1B3 transporters. Omtralo may therefore increase the systemic exposure of OATP1B1 and OATP1B3 substrates such as statins. Co-administration of Omtralo increased the Cmax of atorvastatin and its metabolites by up to 2-fold and AUC by up to 1.3-fold. Therefore, caution should be exercised upon co-administration of Omtralo with statins.
Sildenafil.
Addition of a single dose of sildenafil to Omtralo at steady state in patients with hypertension was associated with greater BP reduction compared to administration of Omtralo alone. Therefore, caution should be exercised when sildenafil or another PDE-5 inhibitor is initiated in patients treated with Omtralo.
Anticipated interactions to be considered.
Potassium.
Concomitant use of potassium-sparing diuretics (e.g. triamterene, amiloride), mineralocorticoid antagonists (e.g. spironolactone, eplerenone), potassium supplements, or salt substitutes containing potassium may lead to increases in serum potassium, and to increases in serum creatinine. Monitoring of serum potassium is recommended if Omtralo is co-administered with these agents (see Section 4.4 Special Warnings and Precautions for Use).
Non-steroidal anti-inflammatory agents (NSAIDs) including selective cyclooxygenase-2 inhibitors (COX-2 inhibitors).
In elderly patients, volume-depleted patients (including those on diuretic therapy), or patients with compromised renal function, concomitant use of Omtralo and NSAIDs may lead to an increased risk of worsening of renal function. Therefore, monitoring of renal function is recommended when initiating or modifying the treatment in patients on Omtralo who are taking NSAIDs concomitantly.
Lithium.
The potential for a drug interaction between Omtralo and lithium has not been investigated. Reversible increases in serum lithium concentrations and toxicity have been reported during concomitant administration of lithium with ACE inhibitors or angiotensin II receptor antagonists. Therefore, careful monitoring of serum lithium levels is recommended during concomitant use with Omtralo. If a diuretic is also used, the risk of lithium toxicity may be increased further.
Frusemide.
Co-administration of Omtralo and frusemide had no effect on the pharmacokinetics of Omtralo but reduced Cmax and AUC of frusemide by 50% and 28%, respectively. While there was no relevant change in urine volume, the urinary excretion of sodium was reduced within 4 hours and 24 hours after co-administration. The average daily dose of frusemide was unchanged from baseline until the end of the PARADIGM-HF study in patients treated with Omtralo.
Transporters.
The active metabolite of sacubitril (sacubitrilat) and valsartan are OATP1B1, OATP1B3 and OAT3 substrates; valsartan is also a MRP2 substrate. Therefore, co-administration of Omtralo with inhibitors of OATP1B1, OATP1B3, OAT3 (e.g. rifampin, cyclosporine) or MRP2 (e.g. ritonavir) may increase the systemic exposure to sacubitrilat or valsartan, respectively. Exercise appropriate care when initiating or ending concomitant treatment with such drugs.
Metformin.
Co-administration of Omtralo with metformin reduced both Cmax and AUC of metformin by 23%. The clinical relevance of these findings is unknown. Therefore, when initiating therapy with Omtralo in patients receiving metformin, the clinical status of the patient should be evaluated.
No significant interactions.
No clinically meaningful drug-drug interaction was observed upon co-administration of Omtralo and digoxin, warfarin, hydrochlorothiazide, amlodipine, omeprazole, carvedilol, intravenous nitroglycerin or a combination of levonorgestrel/ethinyloestradiol. No interaction is expected with atenolol, indomethacin, glyburide, or cimetidine.
CYP 450 interactions.
In vitro metabolism studies indicate that the potential for CYP 450-based drug interactions is low since there is limited metabolism of Omtralo via the CYP450 enzymes. Omtralo does not induce or inhibit CYP450 enzymes.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no available data on the effect of Omtralo on human fertility.
Omtralo did not show any effects on fertility or early embryonic development in male and female rats up to a dose of 73 mg sacubitril/77 mg valsartan/kg/day (≤ 1.0 fold and ≤ 0.13 fold the MRHD on the basis of valsartan and sacubitrilat AUC, respectively). (Category D)
Drugs that act on the renin-angiotensin-aldosterone system (RAAS) can cause fetal and neonatal morbidity and death when administered to pregnant women. Several dozen cases have been reported in the world literature in patients who were taking angiotensin converting enzyme inhibitors (a specific class of drugs acting on the RAAS).
As for other drugs that also act directly on the RAAS, Omtralo must not be used during pregnancy (see Section 4.3 Contraindications) or in women planning to become pregnant. Valsartan exerts its effects via angiotensin II antagonism. There have been reports of injury to the developing fetus (e.g. spontaneous abortion, oligohydramnios and newborn renal dysfunction), when pregnant women have taken valsartan. Physicians prescribing any agents acting on the RAAS should counsel women of childbearing potential about the potential risk of these agents during pregnancy. Patients should be advised to discontinue Omtralo as soon as pregnancies occur and to inform their physicians.
The use of drugs that act directly on the renin-angiotensin-aldosterone system (RAAS) during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function. Oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation and hypoplastic lung development. Prematurity, intrauterine growth retardation and patent ductus arteriosus have also been reported, although it is not clear whether these occurrences were due to exposure to the drug. In addition, in retrospective data, first trimester use of ACE inhibitors has been associated with a potential risk of birth defects.
Infants with histories of in utero exposure to an angiotensin II receptor antagonist should be closely observed for hypotension, oliguria and hyperkalaemia.
In animal studies, Omtralo treatment during organogenesis resulted in increased embryo-fetal lethality in rats at doses ≥ 49 mg sacubitril/51 mg valsartan/kg/day (≤ 0.06 [sacubitrilat, the active metabolite] and 0.7 [valsartan]-fold the maximum recommended human dose [MRHD] of 97 mg/103 mg twice-daily on the basis of the area under the plasma drug concentration-time curve [AUC]) and rabbits at doses ≥ 5 mg sacubitril/5 mg valsartan/kg/day (2-fold and 0.03-fold the MRHD on the basis of valsartan and sacubitrilat AUC, respectively). Omtralo is teratogenic based on a low incidence of fetal hydrocephaly, associated with maternally toxic doses, which was observed in rabbits at a Omtralo dose of ≥ 5 mg sacubitril/5 mg valsartan/kg/day. The adverse embryo-fetal effects of Omtralo are attributed to the angiotensin receptor antagonist activity.
Sacubitril.
There are no data from the use of sacubitril in pregnant women. Studies in animals have shown reproductive toxicity.
Omtralo.
There are no data from the use of Omtralo in pregnant women. Animal studies with Omtralo have shown reproductive toxicity. It is not known whether Omtralo is excreted in human milk. The components of Omtralo, sacubitril and valsartan, were excreted in the milk of lactating rats.
Pre- and postnatal development studies in rats at sacubitril doses up to 750 mg/kg/day (1.1-fold the MRHD on the basis of sacubitrilat AUC) and valsartan at doses up to 600 mg/kg/day (0.9-fold the MRHD on the basis of AUC) indicate that treatment with Omtralo during organogenesis, gestation and lactation may affect pup development and survival.
Because of the potential risk for adverse drug reactions in breastfed newborns/infants, Omtralo is not recommended during breastfeeding. A decision should be made whether to abstain from breast-feeding or to discontinue Omtralo while breast-feeding, taking into account the importance of Omtralo to the mother.
Females of child-bearing potential.
Female patients of child-bearing potential should be advised about the consequences of exposure to Omtralo during pregnancy and to use contraception during treatment with Omtralo and for 1 week after their last dose.
4.7 Effects on Ability to Drive and Use Machines
When driving vehicles or operating machines it should be taken into account that occasionally dizziness or fatigue may occur.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
Summary of the safety profile.
PARADIGM-HF.
The safety of Omtralo in patients with chronic heart failure was evaluated in the pivotal phase 3 study PARADIGM-HF, which compared patients treated twice daily with Omtralo 97 mg/103 mg (n = 4203) or enalapril 10 mg (n = 4229). Patients randomised to Omtralo received treatment for up to 4.3 years, with a median duration of exposure of 24 months; 3271 patients were treated for more than one year.
In the PARADIGM-HF study, patients were previously treated with ACE inhibitors and/or ARBs and also had to successfully complete sequential enalapril and Omtralo run-in periods (median drug exposure of 15 and 29 days, respectively) prior to the randomised double-blind period. During the enalapril run-in period, 1,102 patients (10.5%) permanently discontinued from the study, 5.6% because of an adverse reaction, most commonly renal dysfunction (1.7%), hyperkalaemia (1.7%) and hypotension (1.4%). During the Omtralo run-in period, 10.4% of patients permanently discontinued, 5.9% because of an adverse reaction, most commonly renal dysfunction (1.8%), hypotension (1.7%) and hyperkalaemia (1.3%). Due to discontinuations during the run-in period, the adverse reaction rates as presented in table below may be lower than the adverse reaction rates expected in clinical practice.
Discontinuation of therapy due to an AE in the double-blind period of the PARADIGM-HF trial occurred in 450 (10.71%) of Omtralo treated patients and 516 (12.20%) of patients receiving enalapril. The events most commonly associated with dosage adjustment or treatment interruption were hypotension, hyperkalaemia and renal impairment. The overall incidence of adverse drug reactions (ADRs) of Omtralo in heart failure patients was comparable to enalapril. The pattern of the ADRs is consistent with the pharmacology of Omtralo and the patients underlying conditions.
The overall frequency of adverse reactions was not related to gender, age, or race.
Tabulated summary of adverse drug reactions from clinical trials.
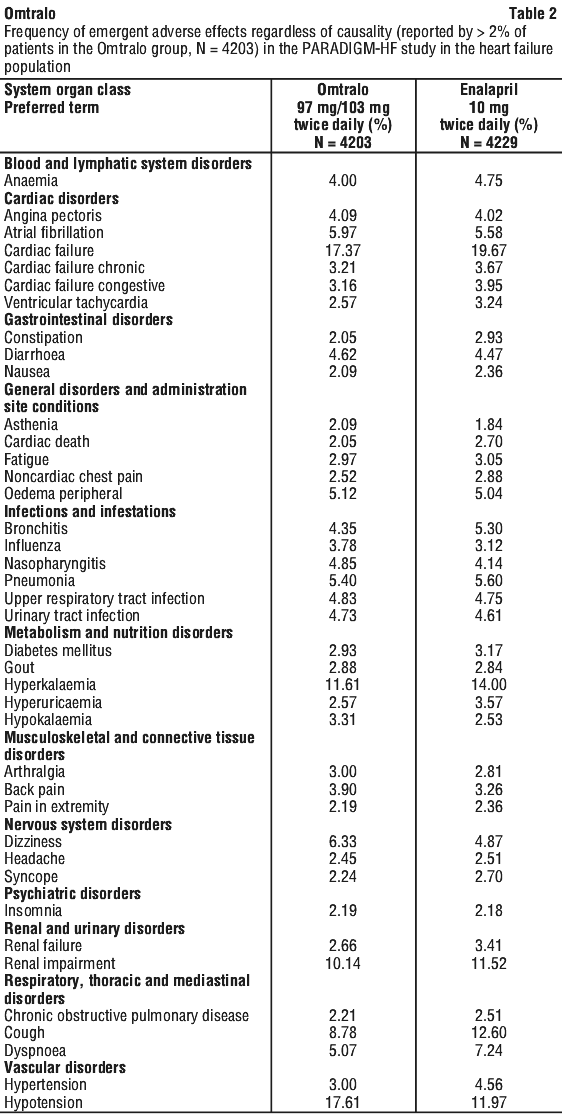
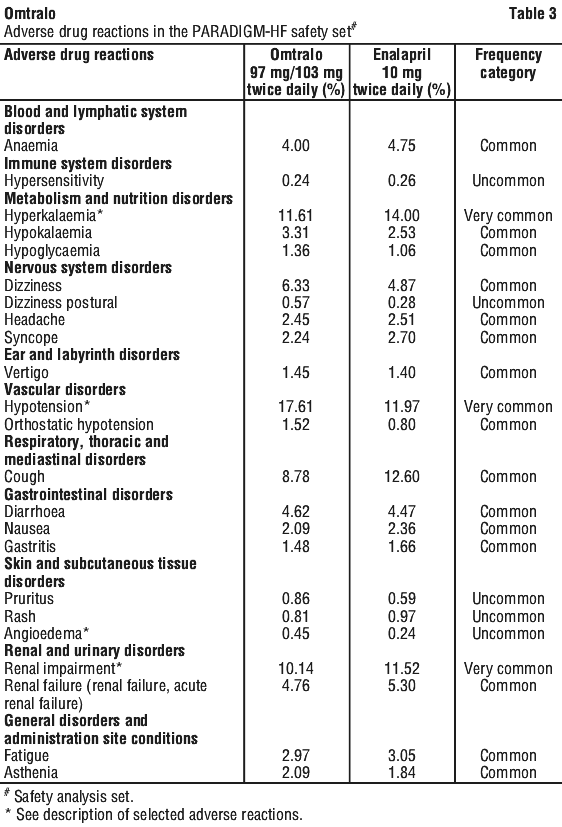
Adverse drug reactions are ranked by system organ class and then by frequency with the most frequent first, using the following convention: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000), including isolated reports. Within each frequency grouping, adverse reactions are ranked in order of decreasing seriousness. See Tables 2 and 3.
Description of selected adverse reactions.
Angioedema.
In the PARADIGM-HF trial, the incidence of angioedema was 0.1% in both the enalapril and Omtralo run-in periods. In the double-blind period, the incidence of angioedema was higher in patients treated with Omtralo than enalapril (0.5% and 0.2%, respectively). The incidence of angioedema in Black patients was 2.4% with Omtralo and 0.5% with enalapril (see Section 4.4 Special Warnings and Precautions for Use).
Hyperkalaemia and serum potassium.
In PARADIGM-HF, hyperkalaemia and serum potassium concentrations > 5.4 mmol/L were reported in 11.6% and 19.7% of Omtralo-treated patients and 14.0% and 21.1% of enalapril-treated patients, respectively.
Blood pressure.
In PARADIGM-HF, hypotension and clinically relevant low systolic blood pressure (< 90 mmHg and decrease from baseline of > 20 mmHg) were reported in 17.6% and 4.76% of Omtralo-treated patients compared with 11.9% and 2.67% of enalapril-treated patients, respectively. Orthostasis was reported in 2.1% of patients treated with Omtralo compared to 1.1% of patients treated with enalapril during the double-blind period of PARADIGM-HF. Falls were reported in 1.9% of patients treated with Omtralo compared to 1.3% of patients treated with enalapril.
Renal impairment.
In PARADIGM-HF, renal impairment was reported in 10.1% of Omtralo-treated patients and 11.5% of enalapril-treated patients.
Laboratory abnormalities.
Hemoglobin and hematocrit.
Decreases in hemoglobin/hematocrit of > 20% were observed in approximately 5% of both Omtralo- and enalapril-treated patients in the double-blind period in PARADIGM-HF.
Serum creatinine.
In PARADIGM-HF, increases in serum creatinine of > 50% were observed in 1.4% of patients in the enalapril run-in period and 2.2% of patients in the Omtralo run-in period. During the double-blind period, approximately 16% of both Omtralo- and enalapril-treated patients had increases in serum creatinine of > 50%.
Serum potassium.
In PARADIGM-HF, potassium concentrations > 5.5 mEq/L were observed in approximately 4% of patients in both the enalapril and Omtralo run-in periods. During the double-blind period, approximately 16% of both Omtralo- and enalapril-treated patients had potassium concentrations > 5.5 mEq/L.
Adverse events leading to study drug discontinuation in the TITRATION study (see Section 5.1 Pharmacodynamic Properties, Clinical trials) are shown in Table 4.
Other Omtralo studies.
PIONEER-HF.
The PIONEER-HF trial was a 8-week study comparing in-hospital initiation of Omtralo versus enalapril in heart failure patients with reduced ejection fraction stabilised following hospitalisation for acute decompensated heart failure (ADHF) (see Section 5.1).
There was no significant difference in the overall occurrence of symptomatic hypotension in patients treated with Omtralo (15.0%) compared to patients treated with enalapril (12.7%). Hyperkalaemia was comparable and did not differ significantly between Omtralo-treated patients (11.6%) and enalapril-treated patients (9.3%). Worsening renal function was reported in 13.6% of Omtralo-treated and 14.7% of enalapril-treated patients. 6 angioedema were reported in the enalapril group (1.4%, all Black patients) compared to 1 in the Omtralo group (0.2%, Caucasian patient).
TRANSITION.
The TRANSITION trial was a 26-week study comparing pre- and post-discharge treatment initiation with Omtralo in 1002 heart failure patients with reduced ejection fraction hospitalised for an acute decompensation event (ADHF) (see Section 5.1).
The safety profile of Omtralo was similar in the pre-discharge and post-discharge groups.
In both studies, the safety profile of Omtralo was consistent with the safety profile in PARADIGM-HF.
Post-marketing experience - adverse drug reactions from spontaneous reports and literature cases (frequency not known).
The following adverse drug reactions have been derived from post-marketing experience with Omtralo via spontaneous case reports and literature cases. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency, which is therefore categorised as not known. Adverse drug reactions are listed according to system organ classes in MedDRA. See Table 5. Additional adverse event information for valsartan may be found in the Australian approved Diovan (valsartan) Product Information.
4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
Limited data are available with regards to over dosage in human subjects with Omtralo. In healthy volunteers, a single dose of Omtralo 583 mg sacubitril/617 mg valsartan, and multiple doses of 437 mg sacubitril/463 mg valsartan (14 days) have been studied and were well tolerated.
Hypotension is the most likely symptom of over dosage due to the blood pressure lowering effects of Omtralo. Symptomatic treatment should be provided.
Omtralo is unlikely to be removed by haemodialysis due to high protein binding.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
The pharmacodynamic effects of Omtralo were evaluated after single and multiple dose administrations in healthy subjects and in patients with heart failure, and are consistent with simultaneous neprilysin inhibition and RAAS blockade. In a 7-day valsartan-controlled study in patients with reduced ejection fraction (HFrEF), administration of Omtralo resulted in a significant non-sustained increase in natriuresis, increased urine cGMP, and decreased plasma MR-proANP and NT-proBNP compared to valsartan. In a 21-day study in HFrEF patients, Omtralo significantly increased urine ANP and cGMP and plasma cGMP, and decreased plasma NT-proBNP, aldosterone and endothelin-1 compared to baseline. Omtralo also blocked the AT1-receptor as evidenced by increased plasma renin activity and plasma renin concentrations. In PARADIGM-HF, Omtralo decreased plasma NT-proBNP and increased plasma BNP and urine cGMP compared with enalapril. While BNP is a neprilysin substrate, NT-proBNP is not. Therefore, NT-proBNP (but not BNP) is a suitable biomarker for monitoring of heart failure patients treated with Omtralo.
In a thorough QTc clinical study in healthy male subjects, single doses of Omtralo 194 mg sacubitril/206 mg valsartan and 583 mg sacubitril/617 mg valsartan had no effect on cardiac repolarisation.
In the mechanistic study PROVE-HF, Omtralo demonstrated improvement of echocardiographic parameters of cardiac structural and functional remodeling (reduction of left atrial volume index [LAVi], left ventricular end systolic [LVESVi] and diastolic volume [LVEDVi] indices, mitral E/E' ratio and increase in left ventricular ejection fraction [LVEF]) compared to baseline at 1 year. Treatment with Omtralo resulted in a clinically meaningful average LVEF increase of 9.4% compared to baseline after 12 months. Omtralo also demonstrated improvement of biomarker parameters (reduction in NT-proBNP, high sensitivity Troponin T and soluble ST2), reflective of an improvement of the underlying pathophysiology of heart failure.
Neprilysin is one of multiple enzymes involved in the clearance of amyloid-β (Aβ) from the brain and cerebrospinal fluid (CSF). Administration of Omtralo 194 mg sacubitril/206 mg valsartan once daily for 2 weeks to healthy subjects was associated with an increase in CSF Aβ 1-38 compared to placebo; there were no changes in concentrations of CSF Aβ 1-40 and 1-42. The clinical relevance of this finding is unknown.
Mechanism of action.
Omtralo exhibits the novel mechanism of action of an angiotensin receptor neprilysin inhibitor (ARNI) by simultaneously inhibiting neprilysin (neutral endopeptidase; NEP) via LBQ657 (sacubitrilat), the active metabolite of the prodrug sacubitril, and by blocking the angiotensin II type-1 (AT1) receptor via valsartan. The complementary cardiovascular benefits and renal effects of Omtralo in heart failure patients are attributed to the enhancement of peptides that are degraded by neprilysin, such as natriuretic peptides (NP), by sacubitrilat and the simultaneous inhibition of the deleterious effects of angiotensin II by valsartan. NPs exert their effects by activating membrane-bound guanylyl cyclase-coupled receptors, resulting in increased concentrations of the second messenger cyclic guanosine monophosphate (cGMP), thereby promoting vasodilation, natriuresis and diuresis, increased glomerular filtration rate and renal blood flow, inhibition of renin and aldosterone release, reduction of sympathetic activity, and anti-hypertrophic and anti-fibrotic effects. Sustained activation of the renin-angiotensin-aldosterone system results in vasoconstriction, renal sodium and fluid retention, activation of cellular growth and proliferation, and subsequent maladaptive cardiovascular remodeling. Valsartan inhibits detrimental cardiovascular and renal effects of angiotensin II by selectively blocking the AT1 receptor, and also inhibits angiotensin II-dependent aldosterone release.
Clinical trials.
Dosing in clinical trials was based on the total amount of both components of Omtralo, i.e. 24 mg/26 mg, 49 mg/51 mg and 97 mg/103 mg were referred to as 50 mg, 100 mg, and 200 mg, respectively.
PARADIGM-HF.
The PARADIGM-HF trial was a multinational, randomised, double-blind trial comparing Omtralo (sacubitril/valsartan) to enalapril in 8,442 adult patients with symptomatic chronic heart failure and reduced ejection fraction (left ventricular ejection fraction ≤ 40% amended later to ≤ 35%), in NYHA class II-IV, in addition to other heart failure therapy. Prior to study enrolment, patients were required to have a plasma B-type natriuretic peptide (BNP) ≥ 150 picogram/mL or N-terminal pro-BNP (NT-proBNP) ≥ 600 picogram/mL, or, if they had been hospitalised for heart failure in the last 12 months, a BNP ≥ 100 picogram/mL or a NT-proBNP ≥ 400 picogram/mL. Patients had to have been on an ACE inhibitor or ARB at a dose equivalent to at least 10 mg of enalapril daily for at least four weeks prior to screening, and on maximally tolerated doses of beta-blockers.
Patients with symptomatic hypotension, or having a systolic blood pressure of < 100 mmHg at screening were excluded. Patients with severe hepatic impairment, eGFR < 30 mL/min/1.73 m2 or serum potassium ≥ 5.2 mmol/L at baseline, or those with any history of angioedema were also excluded. The primary endpoint was the composite of cardiovascular (CV) death or hospitalisation for heart failure.
Prior to study participation, patients were well treated with standard of care therapy which included ACE inhibitors/ARBs (> 99%), beta-blockers (94%), mineralocorticoid antagonists (58%), and diuretics (83%). The median follow-up duration was 27 months and patients were treated for up to 4.3 years.
The population was 66% Caucasian, 18% Asian, and 5% Black; the mean age was 64 years (19% of patients were 75 years or older); and 78% were male. At randomisation, 70% of patients were NYHA class II, 24% were NYHA class III, and 0.7% were NYHA class IV. The mean left ventricular ejection fraction was 29%. There were 11.4% of patients with a baseline left ventricular ejection fraction > 35 and ≤ 40%. Median NTproBNP level at study enrollment was 1,629 picogram/mL for Omtralo-treated patients, and 1,593 picogram/mL for enalapril-treated patients. Median BNP levels at study enrollment was 255 picogram/mL for Omtralo-treated patients, and 251 picogram/mL for enalapril-treated patients. The underlying cause of heart failure was coronary artery disease in 60% of patients; 71% had a history of hypertension, 43% had a history of myocardial infarction, 37% had an eGFR < 60 mL/min/1.73 m2, and 35% had diabetes mellitus. Most patients were taking beta-blockers (94%), mineralocorticoid antagonists (58%), and diuretics (82%). Few patients had an implantable cardioverter-defibrillator (ICD) or cardiac resynchronisation therapy-defibrillator (CRT-D) (15%).
Patients were required to discontinue their existing ACE inhibitor or ARB therapy and entered a sequential single-blind run-in period during which patients received treatment with enalapril 10 mg twice daily, followed by treatment with Omtralo 49 mg/51 mg twice daily, increasing to Omtralo 97 mg/103 mg twice daily. Patients were then randomised to the double-blind period of the study to receive either Omtralo 97 mg/103 mg or enalapril 10 mg twice daily [Omtralo (n = 4209); enalapril (n = 4233)].
In the Omtralo group, 76% of patients remained on the target dose of Omtralo 97 mg/103 mg twice daily at the end of the study (mean daily dose of 375 mg). In the enalapril group, 75% of patients remained on the target dose of 10 mg twice daily at the end of the study (mean daily dose of 18.9 mg).
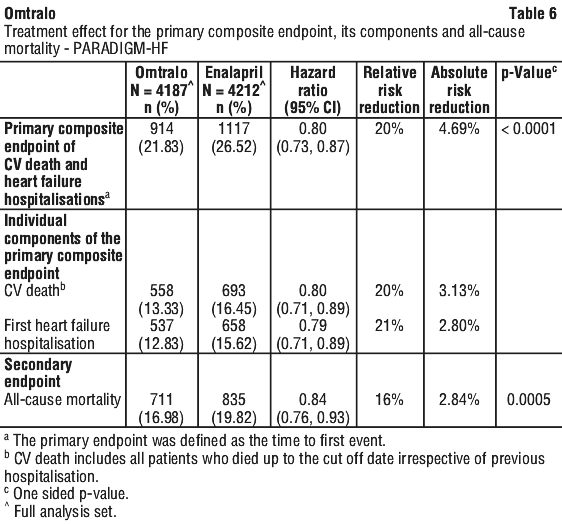
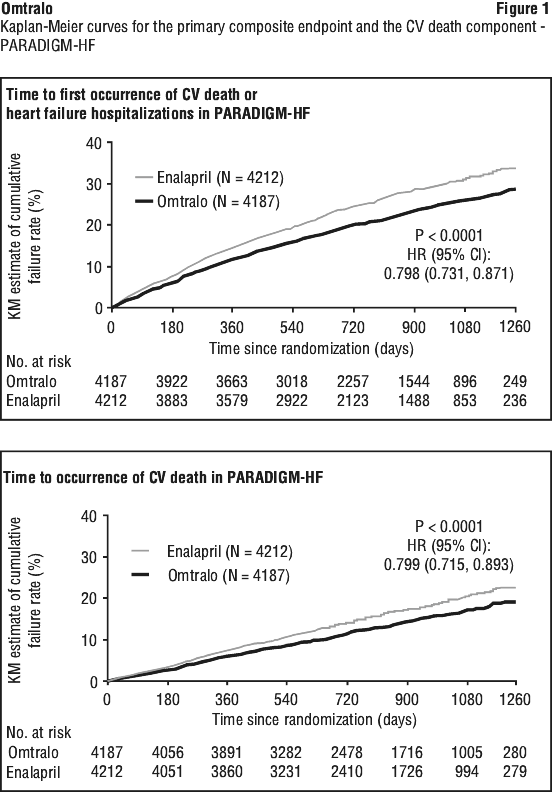
Omtralo demonstrated clinically relevant and statistically significant superiority to enalapril, reducing the risk of cardiovascular death or heart failure hospitalisations by 20% (hazard ratio (HR): 0.80, 95% CI [0.73; 0.87], 1-sided p < 0.0001) versus enalapril. This effect was observed early and was sustained throughout the duration of the trial. The absolute risk reduction was 4.69%. A statistically significant reduction for CV death and first HF hospitalisation was observed (CV death, RRR 20%, HR 0.80; 95% CI [0.71, 0.89]; and hospitalisation for heart failure RRR 21%; HR 0.79; 95% CI [0.71, 0.89]) - see Table 6 and Figure 1. Sudden death accounted for 45% of cardiovascular deaths and was reduced by 20% in Omtralo treated patients compared to enalapril treated patients (HR 0.80). Pump failure accounted for 26% of cardiovascular deaths and was reduced by 21% in Omtralo treated patients compared to enalapril treated patients (HR 0.79).
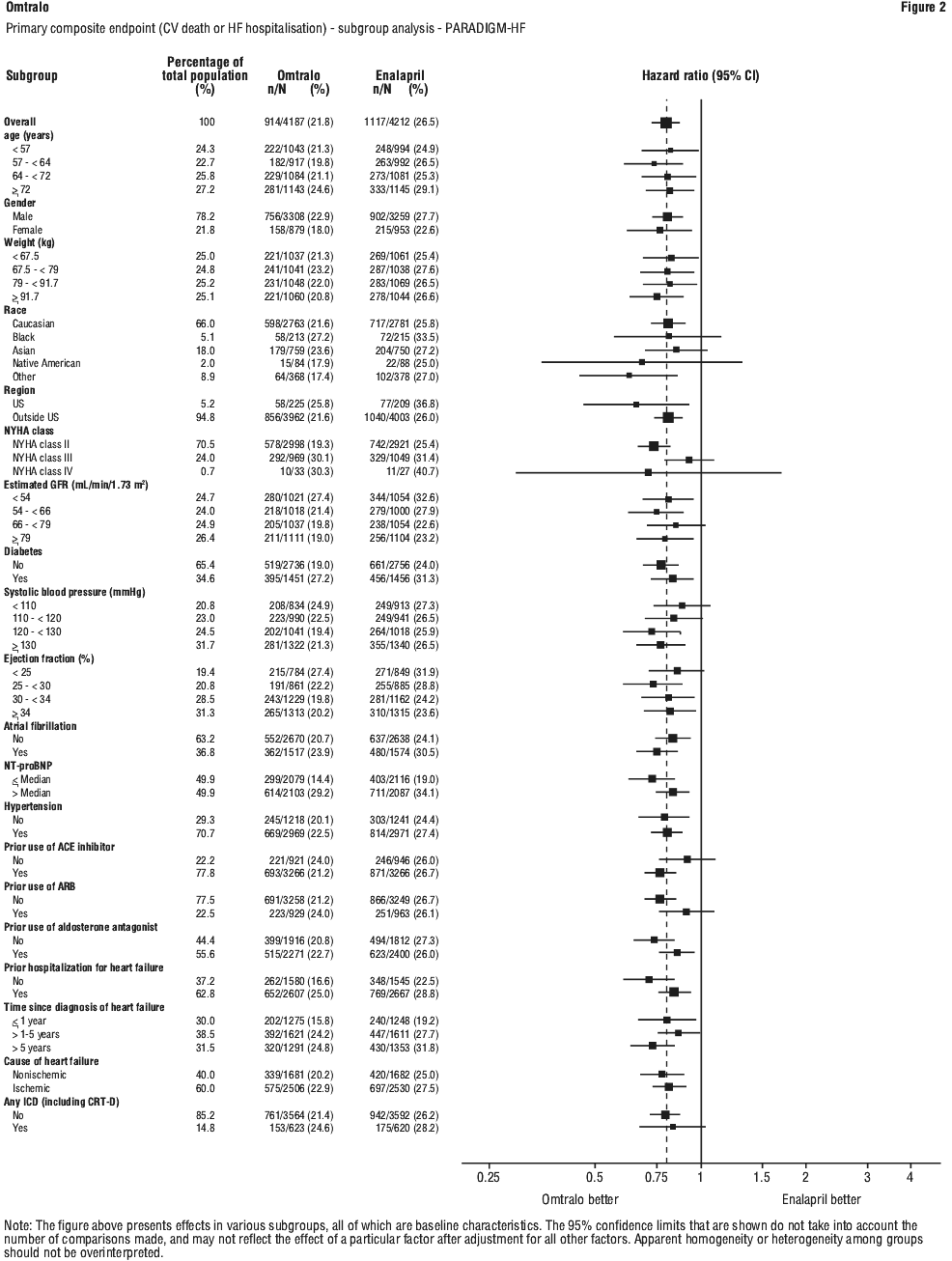
This risk reduction was consistently observed across subgroups including: age, gender, race, geography, NYHA class, ejection fraction, renal function, history of diabetes or hypertension, prior heart failure therapy, and atrial fibrillation.
Omtralo also significantly reduced all-cause mortality by 16% compared with enalapril (RRR 16%, HR 0.84; 95% CI [0.76 to 0.93], 1-sided p=0.0005) - see Table 6. The absolute risk reduction was 2.84%. The Kaplan-Meier presented in Figure 1 (top) shows time to first occurrence of the primary composite endpoint of CV death or heart failure hospitalisation. Omtralo treatment effect was evident early and sustained for the duration of the study. The Kaplan-Meier figure presented in Figure 1 (bottom) shows the time to CV death endpoint. Overall, there were fewer all cause hospital admissions in patients treated with Omtralo compared to enalapril, including a 12% relative risk reduction for the first hospitalisation (HR 0.88 [95% CI: 0.82, 0.94], P < 0.001), and a 16% relative rate reduction for total number of hospitalisations (RR 0.84 [95% CI: 0.78, 0.91], P < 0.001)].
A wide range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes. The results of the primary composite endpoint were consistent across the subgroups examined (Figure 2).
TITRATION.
TITRATION was a 12-week safety and tolerability study in 538 patients with chronic heart failure (NYHA class II - IV) and systolic dysfunction (left ventricular ejection fraction ≤ 35%) naïve to ACE inhibitor or ARB therapy or on varying doses of ACE inhibitors or ARBs prior to study entry. Patients initiated Omtralo 24 mg/26 mg twice daily, were uptitrated to Omtralo 49 mg/51 mg twice daily and then to the target dose of Omtralo 97 mg/103 mg twice daily with either a 3-week or 6-week regimen.
Overall, 76% of patients achieved and maintained the target dose of Omtralo 97 mg/103 mg twice daily without any dose interruption or down-titration over 12-weeks. More patients who were naïve to previous ACE inhibitor or ARB therapy or on low dose therapy (equivalent to < 10 mg of enalapril/day) were able to achieve and maintain Omtralo 97 mg/103 mg when uptitrated over 6 weeks versus 3 weeks.
Other Omtralo studies.
PIONEER-HF.
PIONEER-HF was a 8-week multicenter, randomised, double-blind, parallel group, active-controlled study (n = 881) followed by a 4-week open label period to evaluate safety and tolerability of in-hospital initiation of Omtralo compared to enalapril in HFrEF patients haemodynamically stabilised during hospitalisation for acute decompensated heart failure (ADHF). In PIONEER-HF, 52% of patients were not receiving an ACEi or ARB at the time of admission and 66% had history of HF prior to the index hospitalisation. Omtralo significantly decreased NT-ProBNP vs. enalapril over 8 weeks of treatment (ratio of change with Omtralo vs enalapril, 0.71, p < 0.001). Subgroup analysis of the primary endpoint defined according to demographic and clinical characteristics of interest reflected a consistently beneficial effect of Omtralo compared to enalapril.
TRANSITION.
TRANSITION was a 26-week safety study aimed at exploring whether initiation and up-titration of Omtralo in a wide range of HFrEF patients haemodynamically stabilised after an acute heart failure event, either in-hospital or shortly after discharge, is feasible, safe, and well tolerated. The study was a multicenter, randomised, open label, parallel group study (n = 1002) comparing pre-discharge and post-discharge treatment initiation with Omtralo in heart failure patients with reduced ejection fraction hospitalised for an acute decompensation event (ADHF), due to deterioration of HF (71.1%) or newly diagnosed (de novo) HF (28.9%). The study demonstrated that there were no difference in treatment management between de novo ADHF and deterioration of HF, and between patients with (75.7%) and without (24.3%) prior ACEi or ARB therapy.
In a multivariable analysis, significant (p < 0.05) predictors of target-dose attainment within 10 weeks were age < 65 years, SBP ≥ 120 mmHg at baseline, history of hypertension, de novo HF, no atrial fibrillation at baseline, estimated glomerular filtration rate ≥ 60 mL/min/1.73 m2 at randomisation, and a Omtralo starting dose of 49/51 mg bid. Assignment to pre- or post-discharge initiation of Omtralo was not significant (OR 1.21; 95% CI 0.93-1.59), nor was prior use of an ACEI or ARB a significant predictor of up-titration success (OR 1.04; 95% CI 0.75-1.45).
5.2 Pharmacokinetic Properties
The valsartan contained within Omtralo is more bioavailable than the valsartan in other marketed tablet formulations (see Table 1).
Absorption.
Following oral administration, Omtralo dissociates into sacubitril, which is further metabolised to sacubitrilat, and valsartan, which reach peak plasma concentrations in 0.5 hours, 3 hours, and 1.5 hours, respectively. The oral absolute bioavailability of sacubitril and valsartan is estimated to be ≥ 60% and 23%, respectively.
Following twice daily dosing of Omtralo, steady state levels of sacubitril, sacubitrilat, and valsartan are reached in 3 days. At steady state, sacubitril and valsartan do not accumulate significantly, while sacubitrilat accumulates by 1.6-fold. Omtralo administration with food has no clinically significant impact on the systemic exposures of sacubitril, sacubitrilat and valsartan. Although there is a decrease in exposure to valsartan when Omtralo is administered with food, this decrease is not accompanied by a clinically significant reduction in the therapeutic effect. Omtralo can therefore be administered with or without food.
Distribution.
Omtralo is highly bound to plasma proteins (94% - 97%). Based on the comparison of plasma and CSF exposures, sacubitrilat does cross the blood brain barrier to a limited extent (0.28%). Omtralo has an apparent volume of distribution ranging from 75 L to 103 L.
Metabolism.
Sacubitril is readily converted to sacubitrilat by esterases; sacubitrilat is not further metabolised to a significant extent. Valsartan is minimally metabolised, as only about 20% of the dose is recovered as metabolites. A hydroxyl metabolite has been identified in plasma at low concentrations (< 10%). Since CYP450 enzyme mediated metabolism of sacubitril and valsartan is minimal, co-administration with drugs that impact CYP450 enzymes is not expected to impact the pharmacokinetics.
Excretion.
Following oral administration, 52 - 68% of sacubitril (primarily as sacubitrilat) and ~13% of valsartan and its metabolites are excreted in urine; 37-48% of sacubitril (primarily as sacubitrilat), and 86% of valsartan and its metabolites are excreted in faeces.
Sacubitril, sacubitrilat, and valsartan are eliminated from plasma with a mean elimination half-life (T1/2) of approximately 1.43 hours, 11.48 hours, and 9.90 hours, respectively.
Linearity/ non-linearity.
The pharmacokinetics of sacubitril, sacubitrilat, and valsartan are linear in the dose range tested (24 mg sacubitril/26 mg valsartan - 194 mg sacubitril/206 mg valsartan).
Special populations.
Elderly patients (aged over 65 years).
The exposures of sacubitrilat and valsartan are increased in elderly subjects by 42% and 30%, respectively, compared to younger subjects. However, this is not associated with clinically relevant effects and therefore no dosage adjustment is necessary in patients over 65 years. In patients ≥ 75 years old, a lower starting dose of Omtralo 24 mg/26 mg should be considered (see Section 4.4 Special Warnings and Precautions for Use, Use in the elderly).
Paediatric patients (aged below 18 years).
Omtralo has not been studied in paediatric patients.
Impaired renal function.
A correlation was observed between renal function and systemic exposure to sacubitrilat, but not to valsartan. In patients with mild (60 mL/min/1.73 m2 ≤ eGFR < 90 mL/min/1.73 m2) to moderate (30 mL/min/1.73 m2 ≤ eGFR < 60 mL/min/1.73 m2) renal impairment, the AUC for sacubitrilat was up to 2-fold higher. No dosage adjustment is required in patients with mild or moderate renal impairment. A 2.7-fold higher AUC for sacubitrilat was observed in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2). A starting dose of Omtralo 24 mg/26 mg twice daily is recommended in patients with severe renal impairment. Caution is recommended when administering Omtralo to these patients due to limited data.
No studies have been performed in patients undergoing dialysis. However, sacubitrilat and valsartan are highly bound to plasma protein and, therefore, unlikely to be effectively removed by dialysis.
Impaired hepatic function.
In patients with mild to moderate hepatic impairment, the exposures of sacubitril increased by 1.5- and 3.4-fold, sacubitrilat increased by 1.5- and 1.9-fold, and valsartan increased by 1.2-fold and 2.1-fold, respectively, compared to matching healthy subjects. No dosage adjustment is recommended when administering Omtralo to patients with mild hepatic impairment (Child-Pugh A classification) including patients with biliary obstructive disorders. A starting dose of Omtralo 24 mg/26 mg twice daily is recommended in patients with moderate hepatic impairment (Child-Pugh B classification). Omtralo has not been studied in patients with severe hepatic impairment. Therefore, its use is not recommended in patients with severe hepatic impairment.
Ethnic group.
The pharmacokinetics of Omtralo (sacubitril, sacubitrilat and valsartan) are comparable across different race and ethnic groups (Caucasians, Blacks, Asians, Japanese and others).
Gender.
The pharmacokinetics of Omtralo (sacubitril, sacubitrilat and valsartan) are similar between male and female subjects.
5.3 Preclinical Safety Data
Genotoxicity.
Mutagenicity and clastogenicity studies conducted with Omtralo, sacubitril, and valsartan did not reveal any effects at either the gene or chromosome level.
Carcinogenicity.
Carcinogenicity studies conducted in mice and rats with sacubitril and valsartan did not identify any carcinogenic potential for Omtralo. The doses of sacubitril studied (high dose of 1200 and 400 mg/kg/day in mice and rats, respectively) were about 29 and 19 times, respectively, the maximum recommended human dose (MRHD) on a mg/m2 basis. The doses of valsartan studied (high dose of 160 and 200 mg/kg/day in mice and rats, respectively) were about 4 and 10 times, respectively, the maximum recommended human dose on a mg/m2 basis.
Other preclinical safety findings, including amyloid-β findings.
The effects of Omtralo on amyloid-β concentrations in cerebrospinal fluid (CSF) and brain tissue were assessed in young (2-4 years old) cynomolgus monkeys treated with Omtralo (24 mg sacubitril/26 mg valsartan/kg/day) for 2 weeks. In this study, Omtralo had a pharmacodynamic effect on CSF Aβ clearance in cynomolgus monkeys, increasing CSF Aβ 1-40, 1-42, and 1-38 levels; there was no corresponding increase in Aβ levels in the brain. Increases in CSF Aβ 1-40 and 1-42 were not observed in a 2 week healthy volunteer study in humans (see Section 5.1 Pharmacodynamic Properties). Additionally, in a toxicology study in cynomolgus monkeys treated with Omtralo at 146 mg sacubitril/154 mg valsartan/kg/day for 39-weeks, there was no amyloid-β plaque accumulation in the brain. The clinical relevance of these findings is not known. Studies in heart failure patients will investigate the potential effects of Omtralo on cognitive function and brain amyloid-β deposition.
6 Pharmaceutical Particulars
6.1 List of Excipients
All tablets also contain microcrystalline cellulose, hydroxypropylcellulose, crospovidone, magnesium stearate (vegetable origin), purified talc, colloidal anhydrous silica, hypromellose, titanium dioxide, macrogol 4000 and iron oxide red.
The Omtralo 24/26 and Omtralo 97/103 tablets also contain iron oxide black while the Omtralo 49/51 tablets also contain iron oxide yellow.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C. Protect from moisture.
Store in the original package. Keep out of the reach and sight of children.
6.5 Nature and Contents of Container
PA/Al/PVC/Al blister packs. Blister packs contain 14*, 28* or 56 or 60* tablets.
*Not all packs sizes may be available.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
The sacubitril/valsartan sodium salt complex is a white to almost white powder with a melting point of around 136°C (onset). It is freely soluble in water and the pH is 8.2. The pKas for sacubitril (4.6) and valsartan (3.9 for the carboxylic group, and 4.7 for the tetrazole-NH group) and partition coefficients [sacubitril: log D = 1.29 (n-octanol/phosphate buffer pH 6.8) and valsartan: log D = -1.49 (n-octanol/phosphate buffer pH 7.4)].
Chemical structure.
Active ingredient: a salt complex of the anionic forms of sacubitril and valsartan, sodium cations and water molecules in the molar ratio of 1:1:3:2.5 respectively.
Chemical name (IUPAC): octadecasodium hexakis(4-{[(1S,3R)-1-([1,1'-biphenyl]-4-ylmethyl)-4-ethoxy-3-methyl-4-oxobutyl]amino}-4-oxobutanoate) hexakis(N-pentanoyl-N-{[2'-(1H-tetrazol-1-id-5-yl)[1,1'-biphenyl]-4-yl]methyl}-L-valinate)-water (1/15).
Molecular formula: C288H330N36O48Na18.15H2O.
Relative molecular mass: 5748.0.
CAS number.
936623-90-4.
Following oral administration, the salt complex dissociates into:
Sacubitril (AAN).
Chemical name (IUPAC):4-{[(1S,3R)-1-([1,1'-biphenyl]-4-ylmethyl)-4-ethoxy-3-methyl-4-oxobutyl]amino}-4-oxobutanoic acid.
Molecular formula: C24H29NO5.
Molecular mass: 411.5.
Structural formula:
CAS number.
149709-62-6.
Valsartan (AAN).
Chemical name (IUPAC): N-pentanoyl-N-{[2'-(1H-tetrazol-5-yl)[1,1'-biphenyl]-4-yl]methyl}-L-valine.
Molecular formula: C24H29N5O3.
Molecular mass: 435.5.
Structural formula:
CAS number.
137862-53-4.
7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine Only.
Date published: 01 August 2025
Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. NPS MedicineWise disclaims all liability (including for negligence) for any loss, damage or injury resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.