1 Name of Medicine
Nivolumab/relatlimab.
2 Qualitative and Quantitative Composition
Opdualag 240 mg nivolumab/80 mg relatlimab concentrate for solution for infusion.
Each mL of concentrate for solution for infusion contains 12 mg nivolumab and 4 mg relatlimab.
One vial of 20 mL contains 240 mg nivolumab and 80 mg relatlimab.
Relatlimab (rch) and nivolumab (rch) are human immunoglobulin G4 (IgG4) monoclonal antibodies (HuMAb) produced in Chinese hamster ovary cells by recombinant DNA technology.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrate for solution for infusion (sterile concentrate).
Clear to opalescent, colourless to slightly yellow liquid that is essentially free of particles.
The solution has a pH of approximately 5.8 and an osmolality of approximately 310 mOsm/kg.
4.1 Therapeutic Indications
Opdualag is indicated for the treatment of patients with unresectable or metastatic melanoma who are at least 12 years old.
4.2 Dose and Method of Administration
Treatment must be initiated and supervised by physicians experienced in the treatment of cancer.
The recommended dose of Opdualag for adult patients, or for paediatric patients who are 12 years or older and weigh at least 40 kg, is 480 mg nivolumab and 160 mg relatlimab administered as a 30-minute intravenous infusion once every 4 weeks, until disease progression or unacceptable toxicity.
Dose escalation or reduction is not recommended. In general, withhold Opdualag for severe (Grade 3) immune-related adverse reactions. Permanently discontinue Opdualag for life-threatening (Grade 4) immune-related adverse reactions, recurrent severe (Grade 3) immune-related adverse reactions that require systemic immunosuppressive treatment, or an inability to reduce corticosteroid dose to 10 mg or less of prednisone or equivalent per day within 12 weeks of initiating steroids. Dosage modifications for adverse reactions that require management different from these general guidelines are summarised in Table 1. Detailed guidelines for the management of immune-related adverse reactions are described in Section 4.4 Special Warnings and Precautions for Use.
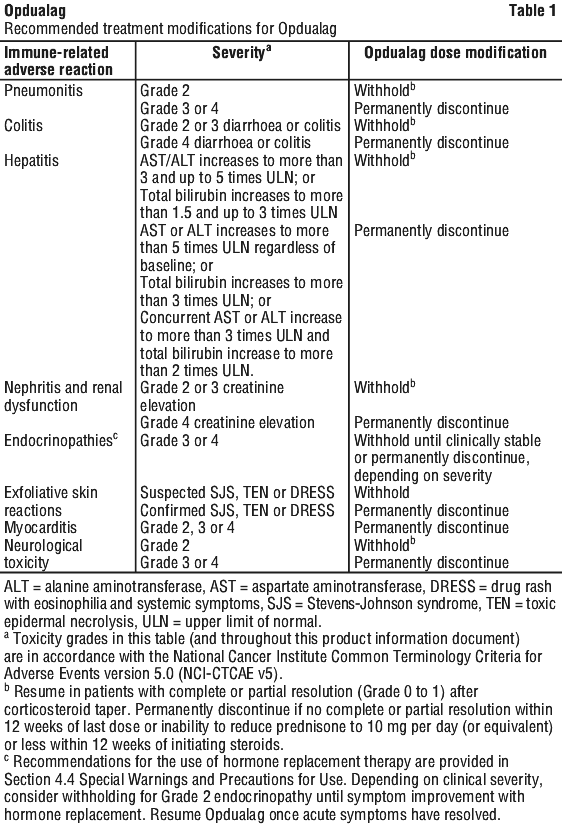
Dose in special populations.
Paediatric patients.
The recommended Opdualag dosage for paediatric patients who are at least 12 years old and weigh at least 40 kg is the same as for adults. A recommended dose has not been established for paediatric patients who are 12 years or older and weigh less than 40 kg.
Elderly patients.
No dose adjustment is required for patients who are at least 65 years old (see Section 5.2 Pharmacokinetic Properties).
Patients with renal impairment.
No dose adjustment is required in patients with mild or moderate renal impairment. The effect of severe renal impairment is unknown (see Section 5.2 Pharmacokinetic Properties).
Patients with hepatic impairment.
No dose adjustment is required in patients with mild or moderate hepatic impairment. The effect of severe hepatic impairment is unknown (see Section 5.2 Pharmacokinetic Properties).
Method of administration.
Opdualag is supplied as a single-dose vial and does not contain any preservatives: aseptic technique must be used during preparation.
Opdualag can be administered either undiluted or after dilution (see Preparing the infusion).
Preparing the infusion. Inspect the Opdualag concentrate for particulate matter or discolouration. Do not shake the vial. Opdualag is a clear to opalescent, colourless to slightly yellow solution. Discard the vial if the solution is cloudy, is discoloured, or contains extraneous particulate matter other than a few translucent to white particles.
Withdraw the required volume of Opdualag concentrate using a sterile syringe and transfer the concentrate into a sterile, intravenous container. Opdualag is compatible with ethylvinyl acetate (EVA), di(2-ethylhexyl) phthalate (DEHP)-plasticised polyvinyl chloride (PVC), and polyolefin (PO) containers.
If diluting prior to administration.
Dilute Opdualag solution with either sodium chloride 9 mg/mL (0.9%) solution for injection or glucose 50 mg/mL (5%) solution for injection to prepare an infusion meeting the following parameters: the final concentration should not be below 3 mg/mL nivolumab and 1 mg/mL relatlimab; the total infusion volume must not exceed 160 mL, or for adult patients weighing less than 40 kg, the total infusion volume should not exceed 4 mL per kilogram of patient weight.
Gently mix the infusion by manual rotation. Do not shake.
Administration. Administer the Opdualag infusion over 30 minutes through an intravenous line containing a sterile, non-pyrogenic, low protein-binding filter (pore size of 0.2 micrometer to 1.2 micrometer).
Do not administer by intravenous push or bolus injection.
Do not co-administer other medicinal products through the same infusion line.
Flush the intravenous line at the end of infusion.
Opdualag is compatible with PVC infusion sets and in-line filters with polyethersulfone (PES), nylon, and polyvinylidene fluoride (PVDF) membranes.4.3 Contraindications
Hypersensitivity to the active substances or any of the excipients listed in Section 6.1.
4.4 Special Warnings and Precautions for Use
Severe and fatal immune-related adverse reactions.
Opdualag can cause immune-related adverse reactions, which can be severe or fatal, and can occur in any organ system or tissue. Immune-related adverse reactions affecting more than one system/tissue can occur simultaneously. Most immune-related adverse reactions manifest during treatment, however, patients should continue to be monitored for a minimum of 5 months after the last dose, as an immune-related adverse reaction to Opdualag may occur at any time during or after discontinuation of therapy.
Early identification and management of immune-related adverse reactions are essential to minimise risk. Monitor patients closely for symptoms and signs that may be clinical manifestations of underlying immune-related adverse reactions. Evaluate hepatic enzymes, creatinine, serum glucose, and thyroid function at baseline and periodically during treatment. Where an immune-related adverse reaction is suspected, conduct investigations to exclude alternative aetiologies, including infection. Institute medical management promptly, including specialist consultation as appropriate. Withhold or discontinue Opdualag based on the severity of a reaction (see Section 4.2 Dose and Method of Administration). In general, if Opdualag requires interruption or discontinuation for an immune-related adverse reaction, commence systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent). Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month to avoid rebound/recurrence. Consider other systemic immunosuppressants if there is worsening or no improvement despite corticosteroid use. Do not resume Opdualag, and consider antibiotic prophylaxis to prevent opportunistic infection, while the patient is receiving immunosuppressive doses of corticosteroids or other immunosuppressive therapy.
Management guidelines for some specific immune-related adverse reactions (some of which may not require systemic steroids) are discussed below.
Immune-related pneumonitis. Opdualag can cause immune-related pneumonitis (interstitial lung disease), and a fatal case occurred in clinical studies (see Section 4.8 Adverse Effects (Undesirable Effects)). Withhold or permanently discontinue Opdualag based on severity (see Section 4.2 Dose and Method of Administration, Table 1). For Grade 3 or 4 pneumonitis, corticosteroids should be initiated at a dose of 2 to 4 mg/kg/day methylprednisolone equivalents. For Grade 2 pneumonitis, corticosteroids may be initiated at a dose of 1 mg/kg/day methylprednisolone equivalents, and increased to 2 to 4 mg/kg/day methylprednisolone equivalents if worsening or no improvement occurs.
Immune-related colitis. Opdualag can cause immune-related colitis, defined as requiring use of corticosteroids and with no clear alternate aetiology (see Section 4.8 Adverse Effects (Undesirable Effects)). Withhold or permanently discontinue Opdualag based on severity (see Section 4.2 Dose and Method of Administration, Table 1).
For Grade 3 or 4 diarrhoea/colitis, corticosteroids should be initiated at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents. For Grade 2 diarrhoea/colitis, corticosteroids may be initiated at a dose of 0.5 to 1 mg/kg/day methylprednisolone equivalents, and increased to 1 to 2 mg/kg/day methylprednisolone equivalents if worsening or no improvement occurs.
Where immune-related colitis appears to be corticosteroid-refractory, consider repeating investigations for infectious diseases including cytomegalovirus (CMV) infection/reactivation. If diagnosis of corticosteroid-refractory immune-related colitis is confirmed, consider alternative immunosuppressive therapy.
Immune-related hepatitis. Opdualag can cause immune-related hepatitis, defined as requiring use of corticosteroids and with no clear alternate aetiology (see Section 4.8 Adverse Effects (Undesirable Effects)). Withhold or permanently discontinue Opdualag based on severity (see Section 4.2 Dose and Method of Administration, Table 1).
For AST or ALT increases to more than 5 times the upper limit of normal (ULN), regardless of baseline, or for total bilirubin increases to more than 3 times ULN, or for AST or ALT increase to more than 3 times ULN with concurrent total bilirubin increase to more than 2 times ULN, corticosteroids should be initiated at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents. For AST/ALT increases to more than 3 but not more than 5 times ULN, or total bilirubin increases to more than 1.5 but not more than 3 times ULN, corticosteroids may be initiated at a dose of 0.5 to 1 mg/kg/day methylprednisolone equivalents and increased to 1 to 2 mg/kg/day methylprednisolone equivalents if liver function is worsening or no improvement occurs.
Immune-related nephritis with renal dysfunction. Opdualag can cause immune-related nephritis with renal dysfunction, defined as requiring use of corticosteroids and with no clear alternate aetiology (see Section 4.8 Adverse Effects (Undesirable Effects)). Withhold or permanently discontinue Opdualag based on severity (see Section 4.2 Dose and Method of Administration, Table 1).
For Grade 4 serum creatinine elevation, corticosteroids should be initiated at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents. For Grade 2 or 3 serum creatinine elevation, corticosteroids may be initiated at a dose of 0.5 to 1 mg/kg/day methylprednisolone equivalents and increased to 1 to 2 mg/kg/day methylprednisolone equivalents if worsening or no improvement occurs.
Immune-related endocrinopathies. Opdualag can cause immune-related endocrinopathies including thyroiditis, hypothyroidism, hyperthyroidism, adrenal insufficiency (primary or secondary), hypophysitis with or without hypopituitarism, and diabetes mellitus with the potential for diabetic ketoacidosis (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients may present with fatigue, headache, mental status changes, abdominal pain, unusual bowel habits, and hypotension, or nonspecific symptoms which may resemble other causes such as brain metastasis or underlying disease.
Unless an alternate aetiology has been identified, signs or symptoms of endocrinopathies should be considered immune-related. Withhold or permanently discontinue Opdualag based on severity (see Section 4.2 Dose and Method of Administration, Table 1).
For symptomatic hypothyroidism, Opdualag should be withheld, and thyroid hormone replacement should be initiated as needed. For symptomatic hyperthyroidism, Opdualag should be withheld and antithyroid medication should be initiated as needed. Corticosteroids at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents should also be considered if acute inflammation of the thyroid is suspected. Upon improvement, Opdualag may be resumed after corticosteroid taper, if needed. Monitoring of thyroid function should continue to ensure appropriate hormone replacement is utilised. Opdualag must be permanently discontinued for life-threatening (Grade 4) hyperthyroidism or hypothyroidism.
Opdualag must be permanently discontinued for severe (Grade 3) or life-threatening (Grade 4) adrenal insufficiency. For symptomatic Grade 2 adrenal insufficiency, Opdualag should be withheld, and physiologic corticosteroid replacement should be initiated as needed. Monitoring of adrenal function and hormone levels should continue to ensure appropriate corticosteroid replacement is utilised.
Opdualag must be permanently discontinued for life-threatening (Grade 4) hypophysitis. For symptomatic Grade 2 or 3 hypophysitis, Opdualag should be withheld, and hormone replacement should be initiated as needed. Corticosteroids at a dose of 1 to 2 mg/kg/day methylprednisolone equivalents should also be considered if acute inflammation of the pituitary gland is suspected. Upon improvement, Opdualag may be resumed after corticosteroid taper, if needed. Monitoring of pituitary function and hormone levels should continue to ensure appropriate hormone replacement is utilised.
For symptomatic diabetes, Opdualag should be withheld, and insulin replacement should be initiated as needed. Monitoring of blood sugar should continue to ensure appropriate insulin replacement is utilised. Opdualag must be permanently discontinued for life-threatening diabetes.
Patients should be monitored for clinical signs and symptoms of endocrinopathies, and for hyperglycaemia and changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation).
Immune-related skin reactions. Opdualag can cause immune-related skin reactions, defined as requiring use of corticosteroids and with no clear alternate aetiology (see Section 4.8 Adverse Effects (Undesirable Effects)). Cases of exfoliative dermatitis, including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug rash with eosinophilia and systemic symptoms (DRESS) have occurred with the use of other therapeutic goods containing an anti-PD-(L)1 antibody such as nivolumab. These are rare, but can be fatal. Withhold or permanently discontinue Opdualag depending on severity (see Section 4.2 Dose and Method of Administration, Table 1).
Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate non-exfoliative rashes. Severe rash should be managed with high-dose corticosteroid (1 to 2 mg/kg/day methylprednisolone equivalents). If exfoliative dermatitis is suspected, refer to a specialised unit for assessment and treatment.
Immune-related myocarditis. Opdualag can cause immune-related myocarditis (see Section 4.8 Adverse Effects (Undesirable Effects)). Diagnosis requires a high index of suspicion. Assess patients with cardiac or cardiopulmonary symptoms for potential myocarditis. If myocarditis is suspected, promptly initiate high dose corticosteroid (1 to 2 mg/kg/day methylprednisolone equivalents) and urgent cardiology consultation with diagnostic workup according to current clinical guidelines. If a diagnosis of myocarditis (Grade 2-4) is established, permanently discontinue Opdualag (see Section 4.2 Dose and Method of Administration, Table 1). If worsening or no improvement occurs despite corticosteroids, corticosteroid dose should be increased to 2 to 4 mg/kg/day methylprednisolone equivalents.
Other immune-related adverse reactions. The following clinically significant immune-related adverse reactions occurred at an incidence of < 1% (unless otherwise noted) in patients who received Opdualag or were reported with the use of other therapeutic goods containing an anti-PD-(L)-1 antibody. Severe or fatal cases have been reported for some of these adverse reactions.
Cardiac/vascular.
Pericarditis, vasculitis.
Nervous system.
Meningitis, encephalitis, myelitis and demyelination, myasthenic syndrome/myasthenia gravis (including exacerbation), myocarditis-myositis-myasthenia gravis overlap syndrome, Guillain-Barré syndrome, nerve paresis, autoimmune neuropathy.
Ocular.
Uveitis, iritis, retinal detachment, visual impairment, blindness. Note: if uveitis occurs in combination with other immune-related adverse reactions, consider a Vogt-Koyanagi-Harada (VKH)-like syndrome, as this may require treatment with systemic steroids to reduce the risk of permanent vision loss.
Gastrointestinal.
Pancreatitis including increases in serum amylase and lipase levels, gastritis, duodenitis.
Musculoskeletal.
Myositis/polymyositis, rhabdomyolysis (and associated sequelae including renal failure), arthritis, polymyalgia rheumatica.
Endocrine.
Hypoparathyroidism.
Other.
Haemolytic anaemia, aplastic anaemia, haemophagocytic lymphohistiocytosis (HLH), systemic inflammatory response syndrome, histiocytic necrotising lymphadenitis (Kikuchi lymphadenitis), sarcoidosis, immune thrombocytopenic purpura, solid organ transplant rejection.
Myocarditis-myositis-myasthenia gravis overlap syndrome.
Cases of myocarditis-myositis-myasthenia gravis overlap syndrome (presenting as an overlap of either two or all three conditions), some with fatal outcome, have been reported with nivolumab in combination with relatlimab. Early recognition and aggressive management are essential to address associated morbidity and risk of mortality.
Patients with pre-existing autoimmune disease (AID).
In patients with pre-existing autoimmune disease (AID), data from observational studies suggest that the risk of immune-mediated adverse reactions following immune checkpoint inhibitor therapy may be increased as compared with the risk in patients without pre-existing AID. In addition, flares of the underlying AID were frequent, but the majority were mild and manageable.
Infusion reactions.
Severe infusion reactions have been reported in clinical trials of nivolumab in combination with relatlimab (see Section 4.8 Adverse Effects (Undesirable Effects)). In case of a severe or life-threatening infusion reaction, Opdualag infusion must be discontinued and appropriate medical therapy administered. Patients with mild or moderate infusion reaction may receive Opdualag with close monitoring and use of premedication according to local treatment guidelines for prophylaxis of infusion reactions.
Complications of allogeneic haematopoietic stem cell transplantation.
Fatal and other serious complications can occur in patients who receive allogeneic haematopoietic stem cell transplantation (HSCT) before or after being treated with a PD-1/PD-L1 inhibitors. Transplant-related complications include hyperacute graft-versus-host-disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause). These complications may occur despite intervening therapy between PD-1/PD-L1 inhibitor treatment and allogeneic HSCT.
Follow patients closely for evidence of transplant-related complications and intervene promptly. Consider the benefit versus risks of treatment with a PD-1/PD-L1 inhibitor prior to or after an allogeneic HSCT.
Patient card.
All prescribers of Opdualag must be familiar with the physician information and management guidelines. The prescriber must discuss the risks of Opdualag therapy with the patient. The patient will be provided with the patient card and instructed to carry the card at all times.
Use in special populations.
Populations excluded from registrational clinical trials.
Patients with active autoimmune disease, medical conditions requiring systemic treatment with moderate or high dose corticosteroids or immunosuppressive medications, uveal melanoma, and active or untreated brain or leptomeningeal metastases were excluded from the pivotal clinical trial of nivolumab in combination with relatlimab. In the absence of data, nivolumab in combination with relatlimab should be used with caution in these populations after careful consideration of the potential benefit/risk on an individual basis.
Patients with renal impairment.
The safety and efficacy of Opdualag have not been studied in patients with severe renal impairment (see Section 5.2 Pharmacokinetic Properties, Special populations).
Patients with hepatic impairment.
The safety and efficacy of Opdualag have not been studied in patients with severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties, Special populations).
Paediatric use.
The safety and efficacy of Opdualag for the treatment of unresectable or metastatic melanoma in paediatric patients who are at least 12 years old and weigh at least 40 kg have been established. This usage is supported by the same evidence that supports this use in adults (see Section 5.1 Pharmacodynamic Properties, Clinical trials), plus additional data analyses that suggest that at the recommended Opdualag dose, nivolumab and relatlimab exposures in paediatric patients 12 years of age who weigh at least 40 kg are expected to result in similar safety and efficacy to that of adults. The pharmacokinetics of monoclonal antibodies and the nature of melanoma is sufficiently similar to allow extrapolation of data from adult patients to pediatric patients 12 years of age or older (who weigh at least 40 kg). A recommended dosage for pediatric patients 12 years of age or older who weigh less than 40 kg has not been established.
The safety and efficacy of Opdualag in children below 12 years of age and/or weighing less than 40 kg have not been established. No data are available.
Elderly patients.
Of 355 patients who received Opdualag in RELATIVITY-047, 47% were 65 years or older, 29% were 65 to 74 years old, 17% were 75 to 84 years old, and 1.7% were 85 years or older. No overall differences in safety or effectiveness were observed between elderly patients and younger patients.4.5 Interactions with Other Medicines and Other Forms of Interactions
Nivolumab and relatlimab are both human monoclonal antibodies and as such, no interaction studies have been conducted. As monoclonal antibodies are not metabolised by cytochrome P450 (CYP) enzymes or other drug metabolising enzymes, inhibition or induction of these enzymes by co-administered medicinal products is not anticipated to affect the pharmacokinetics of nivolumab or relatlimab.
Other forms of interaction.
Systemic immunosuppression.
The use of systemic corticosteroids and other immunosuppressants at baseline (before starting nivolumab in combination with relatlimab) should be avoided because of their potential interference with pharmacodynamic activity. However, systemic corticosteroids and other immunosuppressants can be used after starting nivolumab in combination with relatlimab to treat immune-related adverse reactions.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Studies to evaluate the effect of nivolumab and/or relatlimab on fertility have not been performed, so their effect on fertility (male and female) is unknown.
Contraception.
Advise females of reproductive potential to use effective contraception during treatment and for at least 5 months following the last dose of Opdualag.
(Category D)
Based on its mechanism of action and data from animal studies, Opdualag can cause fetal harm when administered during pregnancy. Administration of nivolumab to cynomolgus monkeys from the onset of organogenesis through delivery resulted in increased abortion and premature infant death (see Animal data). There are no clinical data on the use of Opdualag in pregnancy, however, the PD-1/PD-L1 pathway is involved in maintaining immune tolerance to a fetus. The effects of Opdualag are likely to be greater during the second and third trimesters of pregnancy. Human IgG4 is known to cross the placenta; therefore, nivolumab and relatlimab both have the potential to be transmitted from a pregnant person to the developing fetus. Advise patients of the potential risk to a fetus.
Animal data.
No dedicated reproductive toxicity studies were conducted with Opdualag (nivolumab in combination with relatlimab).
Blockade of the PD-1/PD-L1 pathway has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to increase fetal loss.
The effects of nivolumab on prenatal and postnatal development were evaluated in cynomolgus monkeys that received nivolumab at 10 and 50 mg/kg twice weekly from the onset of organogenesis in the first trimester through delivery, at exposure levels either 8 or 35 times higher than those observed at the clinical dose of 3 mg/kg of nivolumab (based on AUC). There was a dose-dependent increase in fetal losses and increased neonatal mortality beginning in the third trimester and after birth. In surviving infants (18 of 32 compared to 11 of 16 vehicle-exposed infants), no treatment-related clinical signs, alterations to normal development, organ-weight effects, or gross and microscopic pathology changes were seen. Results for growth indices, as well as teratogenic, neurobehavioral, immunological, and clinical pathology parameters throughout the 6-month postnatal period were comparable to the control group. However, based on their mechanism of action, fetal exposure to nivolumab, and, similarly, relatlimab, may increase the risk of developing immune-related disorders or altering the normal immune response. Immune-related disorders have been reported in PD-1 knockout mice and PD-1/LAG-3 knockout mice.
There are no specific animal data on the reproductive toxicity of relatlimab. The effects of a murine surrogate anti-LAG-3 antibody was evaluated in mice using syngeneic and allogeneic breeding models, and no maternal or developmental effects were seen when anti-LAG-3 antibodies were administered beginning on gestation day 6. However, based on the mechanism of action, blockade of LAG-3 with relatlimab can have a similar negative effect as nivolumab on pregnancy.
There are no data on the presence of nivolumab and relatlimab in human milk, the effects on a breastfed child, or the effects on milk production. Because nivolumab and relatlimab may be excreted in human milk and because of the potential for serious adverse reactions in a breastfed child, advise patients not to breastfeed during treatment with Opdualag and for at least 5 months after the last dose.4.7 Effects on Ability to Drive and Use Machines
Opdualag may influence the ability to drive and use machines if adverse reactions such as fatigue occur (see Section 4.8 Adverse Effects (Undesirable Effects)). Advise patients to use caution when driving or operating machinery until they know how Opdualag affects them, and to stop driving and using machines if they experience impairing effects such as fatigue or dizziness.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
The safety of Opdualag has been evaluated in RELATIVITY-047 (study CA224047), a randomised (1:1), double-blinded trial in 714 patients with previously untreated metastatic or unresectable melanoma (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Patients received intravenous infusion with either Opdualag (nivolumab 480 mg and relatlimab 160 mg, n=355) or nivolumab 480 mg (n=359), once every 4 weeks, until disease progression or unacceptable toxicity. The median duration of exposure to Opdualag was 6 months (range: 0 to 31 months) and to nivolumab monotherapy was 5 months (range: 0 to 32 months).
The frequencies included below and in Table 2 are based on all reported adverse reactions, regardless of the investigator assessment of causality. The most common adverse reactions (≥ 20%) were musculoskeletal pain (45%), fatigue (39%), rash (28%), pruritus (25%), and diarrhoea (24%). Serious adverse reactions occurred in 36% of patients who received Opdualag: the most common (≥ 1%) were adrenal insufficiency (1.4%), anaemia (1.4%), colitis (1.4%), pneumonia (1.4%), acute myocardial infarction (1.1%), back pain (1.1%), diarrhoea (1.1%), myocarditis (1.1%), and pneumonitis (1.1%). Incidences of Grade 3-5 adverse reactions in patients with advanced (unresectable or metastatic) melanoma were 40% for Opdualag and 31% for nivolumab monotherapy. Three (0.8%) fatal adverse events occurred in patients receiving Opdualag: one case each of haemophagocytic lymphohistiocytosis, acute oedema of the lung, and pneumonitis.
Opdualag was permanently discontinued due to adverse reactions in 18% of patients. No reactions led to discontinuation in ≥ 1% of patients except for myocarditis (1.7%) and pneumonitis (1.4%).
Dosage interruptions due to an adverse reaction occurred in 43% of patients who received Opdualag. Adverse reactions that required dosage interruption in ≥ 2% of patients who received Opdualag were diarrhoea (3.9%), troponin increased (3.9%), AST increased (2.8%), troponin T increased (2.8%), ALT increased (2.3%), arthralgia (2.3%), hypothyroidism (2.3%), anaemia (2%), fatigue (2%), pneumonitis (2%), and rash (2%).
Tabulated summary of adverse reactions.
Table 2 and Table 3 summarise the adverse reactions and laboratory abnormalities, respectively, reported in RELATIVITY-047.
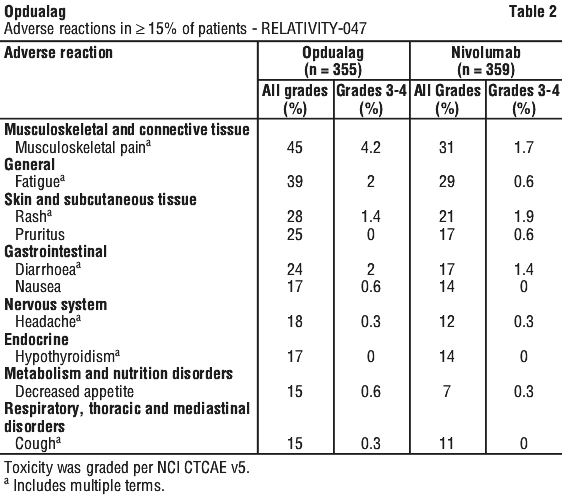 Clinically relevant adverse reactions in < 15% of patients who received Opdualag included vitiligo, adrenal insufficiency, myocarditis, hepatitis and cholangitis.
Clinically relevant adverse reactions in < 15% of patients who received Opdualag included vitiligo, adrenal insufficiency, myocarditis, hepatitis and cholangitis.
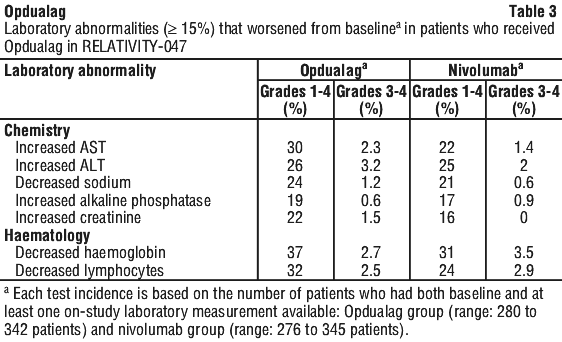
Description of selected immune-related adverse reactions.
Nivolumab and/or relatlimab are associated with immune-related adverse reactions. Most immune-related adverse reactions improved or resolved with appropriate management, including initiation of corticosteroids and treatment modifications, except for endocrinopathies due the continuing need for hormone replacement therapy. The management guidelines for these are described in Section 4.4 Special Warnings and Precautions for Use.
Immune-related pneumonitis.
In patients treated with nivolumab in combination with relatlimab, pneumonitis, including interstitial lung disease and lung infiltration occurred in 4.2% (15/355) of patients. Incidences of Grade 3/4 were 0.8% (3/355). Fatal events occurred in 0.28% (1/355) of patients. Median time to onset was 20 weeks (range: 3.6-94.4). Resolution occurred in 12/15 patients (80.0%) with a median time to resolution of 12.0 weeks (range: 2.3-18.6+). Immune-related pneumonitis led to permanent discontinuation of nivolumab in combination with relatlimab in 1.7% of patients and required high dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) in 60% of patients.
Immune-related colitis.
In patients treated with nivolumab in combination with relatlimab, diarrhoea, colitis, or frequent bowel movements occurred in 14.4% (51/355), of patients. Incidences of Grade 3/4 were 1.7% (6/355). Median time to onset was 11.86 weeks (range: 0.1-95.6). Resolution occurred in 42/50 patients (84.0%) with a median time to resolution of 4.5 weeks (range: 0.1-103.9+). Immune-related colitis led to permanent discontinuation of nivolumab in combination with relatlimab in 2.0% of patients and required high dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) in 35.3% of patients with immune-related colitis.
Immune-related hepatitis.
In patients treated with nivolumab in combination with relatlimab, liver function test abnormalities occurred in 12.1% (43/355) of patients. Incidences of Grade 3/4 were 3.9% (14/355). Median time to onset was 9 weeks (range: 2.0-104.0). Resolution occurred in 34/43 patients (79.1%) with a median time to resolution of 4.71 weeks (range: 0.7-54.0). Immune-related hepatitis led to permanent discontinuation of nivolumab in combination with relatlimab in 1.4% of patients and required high dose corticosteroids in 27.9% of patients with immune-related hepatitis.
Immune-related nephritis or renal dysfunction.
In patients treated with nivolumab in combination with relatlimab, nephritis or renal dysfunction occurred in 3.9% (14/355) of patients. Incidences of Grade 3/4 were 1.4% (5/355). Median time to onset was 18.36 weeks (range: 1.9-98.1). Resolution occurred in 12/14 patients (85.7%) with a median time to resolution of 12.43 weeks (range: 0.9-51.0+). Immune-related nephritis and renal dysfunction led to permanent discontinuation of nivolumab in combination with relatlimab in 1.1% of patients and required high dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) in 21.4% of patients with immune-related nephritis and renal dysfunction.
Immune-related endocrinopathies.
In patients treated with nivolumab in combination with relatlimab, thyroid disorders, including hypothyroidism or hyperthyroidism, occurred in 19.4% (69/355) of patients. There were no incidences of Grade 3/4 thyroid disorder. Incidences of Grade 3/4 adrenal insufficiency occurred in 0.8% (3/355). There were no incidences of Grade 3/4 hypophysitis or hypopituitarism. Incidences of Grade 3/4 diabetes mellitus (including Type 1 diabetes mellitus) were in 0.3% (1/355). Median time to onset of these endocrinopathies was 12.29 weeks (range: 1.0-71.0). Resolution occurred in 20/85 patients (23.5%). Time to resolution ranged from 0.1+-138.1+ weeks. Immune-related endocrinopathies led to permanent discontinuation of nivolumab in combination with relatlimab in 1.1% of patients and required high dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) in 7.1% of patients with immune-related endocrinopathies.
Immune-related skin reactions.
In patients treated with nivolumab in combination with relatlimab, rash occurred in 42.3% (150/355) of patients. Incidences of Grade 3/4 events were 1.4% (5/355). Median time to onset was 6.5 weeks (range: 0.1-97.9). Resolution occurred in 64/150 patients (42.7%). Time to resolution ranged from 0.1-142.6+ weeks. Immune-related skin adverse reactions led to permanent discontinuation of nivolumab in combination with relatlimab in 0.3% of patients and required high dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) in 2.7% of patients with immune-related skin adverse reactions.
Immune-related myocarditis.
In patients treated with nivolumab in combination with relatlimab, myocarditis occurred in 1.4% (5/355) of patients. Incidences of Grade 3/4 events were 0.6% (2/355). Median time to onset was 4.14 weeks (range: 2.1-6.3). Resolution occurred in 5/5 patients (100%) with a median time to resolution of 3 weeks (1.9-14.0). Myocarditis led to permanent discontinuation of nivolumab in combination with relatlimab in 1.4% of patients and required high dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) in 100% of patients with immune-related myocarditis.
Infusion reactions.
In patients treated with nivolumab in combination with relatlimab, hypersensitivity/infusion reactions occurred in 6.5% (23/355) of patients. All incidents were Grade 1/2.
Immunogenicity.
In study CA224047 out of the evaluable patients for anti-drug antibodies, the incidence of treatment-emergent anti-relatlimab antibodies and neutralising antibodies against relatlimab in the Opdualag group were 5.6% (16/286) and 0.3% (1/286), respectively. The incidence of treatment-emergent anti-nivolumab antibodies and neutralising antibodies against nivolumab in the Opdualag group [3.8% (11/288) and 0.3% (1/288), respectively] were similar to that observed in the nivolumab group [5.9% (16/272) and 0.4% (1/272), respectively].
Postmarketing experience.
The events listed below have been reported during the post-approval use of nivolumab and/or its combination with relatlimab. These reports come from voluntary submissions from an unspecified population size, thus preventing the estimation of their frequency.
Musculoskeletal and connective tissue disorders.
Sjogren's syndrome, tenosynovitis.
Nervous system disorders.
Myasthenia gravis, myocarditis-myositis-myasthenia gravis overlap syndrome.
Immune system disorders.
Infusion related reaction (including cytokine release syndrome).
Gastrointestinal disorders.
Coeliac disease, pancreatic insufficiency, enterocolitis.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.4.9 Overdose
No cases of overdose were reported in the pivotal clinical trial (CA224047). In case of overdose, patients should be closely monitored for signs or symptoms of adverse reactions, and appropriate symptomatic treatment instituted immediately.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Relatlimab is a human IgG4 monoclonal antibody that binds to the LAG-3 T cell receptor, blocks interaction with its ligands (including major histocompatibility complex [MHC] class II), and reduces LAG-3 pathway-mediated inhibition of the immune response. Antagonism of this pathway promotes T cell proliferation and cytokine secretion.
Binding of the PD-1 receptor found on T cells to its ligands (PD-L1 and PD-L2), inhibits T cell proliferation and cytokine production. Upregulation of PD-1 ligands occurs in some tumours and signalling through this pathway can contribute to inhibition of active T cell immune surveillance of tumours. Nivolumab is a human IgG4 monoclonal antibody that binds to the PD-1 receptor, blocks interaction with PD-L1 and PD-L2, and reduces PD-1 pathway-mediated inhibition of the immune response, including the anti-tumour immune response. In syngeneic mouse tumour models, blocking PD-1 activity resulted in decreased tumour growth.
Combined inhibition of PD-1 (with nivolumab) and LAG-3 (with relatlimab) results in increased T-cell activation compared to the effect of either antibody alone. In murine syngeneic tumour models, LAG-3 blockade potentiates the anti-tumour activity of PD-1 blockade, inhibiting tumour growth and promoting tumour regression.
Pharmacodynamics.
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of Opdualag have not been fully characterised.
Clinical trials.
Unresectable or metastatic melanoma. Randomised phase 2/3 study of nivolumab in combination with relatlimab vs. nivolumab (CA224047).
The safety and efficacy of Opdualag were investigated in a randomised, double-blinded study (CA224047/RELATIVITY-047) in patients with previously untreated, histologically confirmed stage III (unresectable) or stage IV melanoma per American Joint Committee on Cancer (AJCC) version 8. Patients were allowed to have received prior adjuvant or neoadjuvant melanoma therapy (anti PD-(L)1, anti-CTLA-4, or BRAF/MEK targeted therapy was allowed as long as there was at least 6 months between the last dose of therapy and date of recurrence; interferon therapy was allowed as long as the last dose was at least 6 weeks prior to randomisation). Patients with active autoimmune disease, medical conditions requiring systemic treatment with moderate or high dose corticosteroids or immunosuppressive medications, uveal melanoma, active or untreated brain or leptomeningeal metastases and those with a history of myocarditis, elevated troponin levels > 2 times ULN or ECOG performance status score ≥ 2 were excluded from the study.
A total of 714 patients were randomised to receive either Opdualag (nivolumab 480 mg and relatlimab 160 mg, n=355) or nivolumab 480 mg as monotherapy (n=359) by intravenous infusion once every 4 weeks, until disease progression or unacceptable toxicity. Randomisation was stratified by tumour PD-L1 expression (≥ 1% vs. < 1) according to the 28-8 pharmDx IHC test, LAG-3 expression (≥ 1% vs. < 1) as determined by an analytically validated LAG-3 IHC clinical trial assay, BRAF V600 mutation status, and AJCC M stage (M0/M1any[0] vs. M1any[1]). Tumour assessments, according to the Response Evaluation Criteria in Solid Tumours (RECIST), version 1.1, were conducted 12 weeks after randomisation and continued every 8 weeks up to 52 weeks and then every 12 weeks thereafter. The primary efficacy outcome measure was progression-free survival (PFS) determined by Blinded Independent Central Review (BICR). The secondary efficacy outcome measures were overall survival (OS), and overall response rate (ORR) by BICR.
Amongst the randomised population, the median age was 63 years (range: 20-94), 58% were male, and 97% were white. Baseline ECOG performance status was 0 (67%) or 1 (33%). The majority of the patients had AJCC Stage IV disease (92%); 39% had M1c, 2.4% had M1d disease, and 36% of patients had a baseline LDH level greater than ULN at study entry. Thirty-nine percent of patients had BRAF mutation positive melanoma, 75% had LAG-3 ≥ 1% and 41% of patients had PD-L1 ≥ 1% tumour cell membrane expression.
The primary efficacy endpoint for PFS is shown in Figure 1 and Table 4 and secondary efficacy outcomes (OS and ORR) are also included in Table 4.
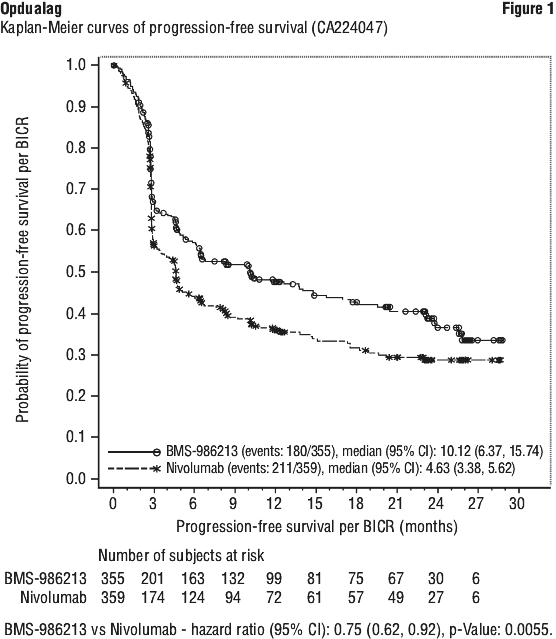
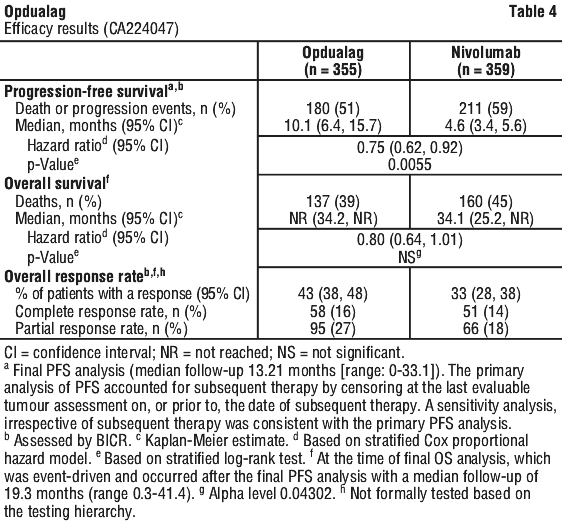 In exploratory analyses, hazard ratio (HR) point estimates for PFS favoured Opdualag across prespecified subgroups, including those defined by the study stratification factors (PD-L1, LAG-3 and BRAF status, and M-stage per AJCC version 8), and key clinical subgroups including baseline ECOG performance status, age, history of brain metastases, and baseline LDH level.
In exploratory analyses, hazard ratio (HR) point estimates for PFS favoured Opdualag across prespecified subgroups, including those defined by the study stratification factors (PD-L1, LAG-3 and BRAF status, and M-stage per AJCC version 8), and key clinical subgroups including baseline ECOG performance status, age, history of brain metastases, and baseline LDH level.
In the PD-L1 < 1% subgroup, the HR (95% CI) for PFS was 0.66 (0.51, 0.84) with median PFS of 6.4 and 2.9 months for the Opdualag and nivolumab arms, respectively. In the PD-L1 ≥ 1% subgroup, the HR for PFS was 0.95 (95% CI: 0.68, 1.33) with median PFS of 15.7 and 14.7 months for the Opdualag and nivolumab arms, respectively.
5.2 Pharmacokinetic Properties
The pharmacokinetics (PK) of relatlimab following its administration in combination with nivolumab was characterised in patients with various cancers who received relatlimab doses of 20 to 800 mg every 2 weeks (0.25 to 10 times the approved recommended dose) or 160 to 1440 mg every 4 weeks (1 to 9 times the approved recommended dose) either as a monotherapy or in combination with nivolumab doses of 80 or 240 mg every 2 weeks or 480 mg every 4 weeks.
Steady-state concentrations of relatlimab were reached by 16 weeks with an every 4 weeks regimen and the systemic accumulation was 1.9-fold. The average concentration (Cavg) of relatlimab after the first dose increased dose proportionally at doses ≥ 160 mg every 4 weeks. See Table 5.
 In CA224047, the nivolumab geometric mean Cmin at steady state was similar between the nivolumab in combination with relatlimab arm and the nivolumab monotherapy arm.
In CA224047, the nivolumab geometric mean Cmin at steady state was similar between the nivolumab in combination with relatlimab arm and the nivolumab monotherapy arm.
Distribution.
The geometric mean value (CV%) for relatlimab volume of distribution at steady state is 6.65 L (19.8%) and nivolumab is 6.65 L (19.2%).
Elimination.
The geometric mean (CV%) clearance of relatlimab is 5.5 mL/h (41%) at steady state: 10% lower than after the first dose [6 mL/h (39%)]. Following administration of nivolumab 480 mg and relatlimab 160 mg administered every 4 weeks, the geometric mean (CV%) effective half-life (t1/2) of relatlimab is 26.2 days (37%).
The geometric mean (CV%) clearance of nivolumab is 7.6 mL/h (40%) at steady state: 21% lower than that after the first dose [9.6 mL/h (40%)] and the terminal half-life (t1/2) is 26.5 days (36%).
Special populations.
A population PK analysis suggested that the following factors had no clinically important effect on the CL of nivolumab and relatlimab: age (range: 17 to 92 years), sex, race, mild or moderate renal impairment (eGFR 30 to 89 mL/min/1.73 m2), and mild hepatic impairment (total bilirubin [TB] less than or equal to upper limit of normal [ULN] and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST) or moderate hepatic impairment (TB greater than 1.5 to 3 times ULN and any AST).
The effects of severe renal or hepatic impairment on the pharmacokinetics of nivolumab and relatlimab are unknown.
The exposures of nivolumab and relatlimab in paediatric patients 12 years of age or older who weigh at least 40 kg are expected to be in the range of exposures in adult patients at the recommended dose.
5.3 Preclinical Safety Data
Genotoxicity.
Studies to evaluate the genotoxic potential of nivolumab or relatlimab have not been performed.
Carcinogenicity.
Studies to evaluate the carcinogenic potential of nivolumab or relatlimab have not been performed.6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, histidine hydrochloride monohydrate, sucrose, pentetic acid (diethylenetriaminepentaacetic acid), polysorbate 80 (E433), water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products. Opdualag should not be infused concomitantly in the same intravenous line with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store refrigerated at 2°C-8°C.
Do not freeze.
Store in the original package in order to protect from light.
The unopened vials can be stored at controlled room temperature up to 25°C with room light for up to 72 hours.
After preparation of the infusion.
This medicinal product does not contain any preservatives.
To reduce microbiological hazard, once opened, the medicinal product should be infused, or diluted and infused, immediately.
If not used immediately, store the diluted solution either:
At room temperature and room light for no more than 8 hours from the time of preparation to end of the infusion. Discard diluted solution if not used within 8 hours from the time of preparation; or
under refrigeration at 2°C to 8°C with protection from light for no more than 24 hours from the time of preparation, which includes the time allowed for equilibration of the infusion bag to room temperature and the duration of the infusion. Discard diluted solution if not used within 24 hours from the time of preparation.
Do not freeze.
6.5 Nature and Contents of Container
Pack of one 25 mL vial (Type I glass), stopper (coated butyl rubber) and a yellow flip-off aluminium seal. Each vial is filled with 21.3 mL of solution, which includes an overfill of 1.3 mL for vial, needle, and syringe holdup.
6.6 Special Precautions for Disposal
Each vial of Opdualag is for single use in one patient only. Discard any residue.
Do not store any unused portion of the infusion solution for reuse.
In Australia, any unused medicinal product or waste material should be discarded in accordance with local requirements.
6.7 Physicochemical Properties
CAS number.
Nivolumab.
CAS: 946414-94-4.
Relatlimab.
CAS: 1673516-98-7.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
