

What is in this leaflet
This leaflet answers some common questions about Otezla. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Otezla against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What OTEZLA is used for
Otezla is used to treat adults with the following conditions:
- Moderate to severe plaque psoriasis (an inflammatory disease of the skin, which can cause red, scaly, thick, itchy, painful patches on your skin, and can also affect your scalp and nails)
- Psoriatic arthritis (an inflammatory disease of the joints, often accompanied by psoriasis)
Psoriasis and psoriatic arthritis are usually lifelong conditions and there is currently no cure. Otezla works by reducing the activity of a natural substance in the body's cells called 'phosphodiesterase 4'. This helps regulate the immune response associated with psoriasis and psoriatic arthritis. By regulating the immune response, Otezla can help to control the signs and symptoms of these conditions.
In psoriasis, treatment with Otezla results in a reduction in psoriatic skin plaques and other signs and symptoms of the disease.
In psoriatic arthritis, treatment with Otezla results in an improvement in swollen and painful joints, and can improve your general physical function.
Otezla has also been shown to improve the quality of life in patients with psoriasis or psoriatic arthritis. This means that the impact of your condition on daily activities, relationships and other factors should be less than it was before.
Ask your doctor if you have any questions about how Otezla works, or why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
Before you take OTEZLA
When you must not take it
Do not take Otezla if you have an allergy to apremilast, the active ingredient of Otezla, or any of the other ingredients of Otezla listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
If you think you may be allergic to Otezla, ask your doctor for advice.
Do not take Otezla if you are pregnant or think you may be pregnant.
Do not breast-feed if you are taking this medicine.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. In that case, return it to your pharmacist.
Before you start to take it
Follow your doctor's instructions carefully.
If you have not fully understood these instructions, ask your doctor again before taking Otezla.
Tell your doctor if you have or have had any kidney problems.
Tell your doctor if you are pregnant, think you may be pregnant or are planning to have a baby. There is little information about the effects of Otezla in pregnancy. You should not use Otezla if you are pregnant. You should not become pregnant while taking this medicine
Tell your doctor before taking this medicine if you are breast-feeding or intent to breast-feed. It is not known whether Otezla passes into the mother's milk. You should not use Otezla while breast-feeding.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have experienced depression or have had suicidal thoughts in the past.
During treatment with Otezla, if you feel depressed, have suicidal thoughts, other mood changes or notice that these events have worsened, tell your doctor as soon as possible.
During treatment with Otezla, if you experience severe diarrhoea, nausea or vomiting, tell your doctor.
Tell your doctor if you have rare hereditary problems to some sugars (e.g., galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption). Otezla contains lactose (a type of sugar). Tell your doctor if you have lactose intolerance.
Do not give this medicine to a child or adolescent under the age of 18 years. Safety and effectiveness of Otezla in children under the age of 18 years have not been established.
If you have not told your doctor about any of the above, tell him/ her before you start taking Otezla.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
In particular, tell your doctor or pharmacist before taking Otezla if you are taking any of the following medicines:
- rifampicin, an antibiotic used for tuberculosis
- carbamazepine, phenytoin and phenobarbitone, medicines used in the treatment of seizures or epilepsy
- St. John's Wort, a herbal medicine for mild anxiety and depression.
These medicines may be affected by Otezla or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking Otezla.
How to take OTEZLA
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
How much to take
Your doctor will tell you how much Otezla to take and for how long you will need to take it. Your doctor will monitor your progress.
- When you first start taking Otezla, you will receive a treatment initiation pack which contains all the doses as listed in the table below.
- This pack is clearly labelled to make sure you take the correct tablet at the correct time.
- Your treatment will start at a lower dose and will gradually be increased (also called 'titrated') over the first 6 days of treatment.
- This initiation pack will also contain enough tablets for another 8 days at the recommended dose (Days 7 to 14).
- The final recommended dose of Otezla is a 30 mg tablet twice a day; one 30 mg dose in the morning and one 30 mg dose in the evening. This is a total daily dose of 60 mg.
- By the end of Day 6 you will have reached this recommended dose.
- Once the recommended dose has been reached, you will only get the 30 mg tablet strength in your prescribed packs.
- You will only ever go through a titration stage once, even if you re-start treatment after a break.
Day 1: total dose - 10 mg
- Morning dose
10 mg - pink tablet - Evening Dose
Do not take a dose
Day 2: total dose - 20 mg
- Morning dose
10 mg - pink tablet - Evening Dose
10 mg - pink tablet
Day 3: total dose - 30 mg
- Morning dose
10 mg - pink tablet - Evening Dose
20 mg - brown tablet
Day 4: total dose - 40 mg
- Morning dose
20 mg - brown tablet - Evening Dose
20 mg - brown tablet
Day 5: total dose - 50 mg
- Morning dose
20 mg - brown tablet - Evening Dose
30 mg - beige tablet
Day 6 onwards: total dose - 60 mg
- Morning dose
30 mg - beige tablet - Evening Dose
30 mg - beige tablet
If you have severe kidney problems, the recommended dose of Otezla is 30 mg once a day. Your doctor will tell you how to gradually increase (titrate) your dose when you first start taking Otezla. Your doctor may advise that you only take the morning dose shown in the table above, and that you skip the afternoon or evening dose.
How to take it
Swallow the tablets whole, preferably with water, either with or without food, twice a day as directed by your doctor.
Do not crush, split or chew the tablets.
When to take it
Take Otezla at about the same time each day, one tablet in the morning and one tablet in the evening.
How long to take it
Your doctor will tell you how long to continue taking Otezla.
If you forget to take it
Take it as soon as you remember. If it is close to the time for the next dose, just skip the missed dose. Take the next dose at the regular time. Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at your nearest hospital, if you think that you or anyone else may have taken too much Otezla.
Do this even if there are no signs of discomfort or poisoning.
While you are using OTEZLA
Things you must do
Tell any other doctors, dentists and pharmacists who are treating you that you are taking Otezla.
If you are about to be started on any new medicine, remind your doctor, dentist or pharmacist that you are taking Otezla.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not breast-feed or become pregnant whilst taking Otezla.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not take Otezla to treat any other complaints unless your doctor tells you to.
Things to be careful of
Be careful driving or operating machinery until you know how Otezla affects you.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Otezla.
Like all medicines, Otezla can have side effects, although not everybody gets them and some are uncommon. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- diarrhoea; nausea; vomiting; stomach pain; indigestion; decrease in appetite
- weight decrease
- cough; cold; runny nose; inflammation/infection of the nose, throat, sinus, or upper respiratory tract
- if you feel tired; if you have trouble or difficulty sleeping
- headaches and migraine headaches
- back pain; joint pain; painful, swollen joints
- Increased blood pressure
- skin rash
- allergic reactions.
The above list mainly includes the more common side effects of your medicine.
Tell your doctor as soon as possible if you notice any of the following:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body.
Tell your doctor or pharmacist immediately if any of the side effects gets worse, or if you notice any other side effects not listed in this leaflet.
After using OTEZLA
Storage
Keep your tablets in a dry place where the temperature stays below 30C.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Medicines should not be disposed of via wastewater or household waste. These measures will help to protect the environment.
Product description
What it looks like
Apremilast 10 mg Tablets: Pink, diamond shaped film-coated tablet with "APR" engraved on one side and "10" on the opposite side.
Apremilast 20 mg Tablets: Brown, diamond shaped film-coated tablet with "APR" engraved on one side and "20" on the opposite side.
Apremilast 30 mg Tablets: Beige, diamond shaped film-coated tablet with "APR" engraved on one side and "30" on the opposite side.
The tablets are provided in packs as follows.
- A titration pack that will last for two weeks. This pack will contain 4 x 10 mg, 4 x 20 mg and 5 x 30 mg tablets for the first week and 14 x 30 mg tablets for the second week.
- A four-week pack (56 x 30 mg tablets).
Ingredients
Otezla tablets contain an active ingredient called apremilast.
The other ingredients are:
- lactose monohydrate
- microcrystalline cellulose
- croscarmellose sodium
- magnesium stearate.
The film coat contains:
Polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, iron oxide red. In addition, iron oxide yellow (in 20 mg and 30 mg tablets only) and iron oxide black (in 30 mg tablets only).
Supplier
Otezla is supplied in Australia by:
Amgen Australia Pty Ltd
ABN 31 051 057 428
Level 7, 123 Epping Road
North Ryde
NSW 2113
Medical Enquiries 1800 803 638
Email: [email protected]
This leaflet was updated in Jan 2021.
Australian Registration Numbers:
Otezla (apremilast) titration pack - AUST R 220424
Otezla (apremilast) 30 mg tablet blister pack - AUST R 220423
Published by MIMS March 2021

 In the Phase 3 study in adults with mild to moderate plaque psoriasis, the safety profile observed in the Otezla group during the placebo controlled phase was overall consistent with the safety profile previously established in adult patients with moderate to severe plaque psoriasis. This safety profile remained similar throughout the study.
In the Phase 3 study in adults with mild to moderate plaque psoriasis, the safety profile observed in the Otezla group during the placebo controlled phase was overall consistent with the safety profile previously established in adult patients with moderate to severe plaque psoriasis. This safety profile remained similar throughout the study. The most commonly reported adverse reactions in Phase 3 (Studies PALACE 1, PALACE 2, PALACE 3, PALACE 4 and ESTEEM 1 and ESTEEM 2) clinical studies have been gastrointestinal (GI) disorders including diarrhoea (15.7%) and nausea (13.9%). These GI adverse reactions were mostly mild to moderate in severity, with 0.3% of patients reporting severe diarrhoea and 0.3% of patients reporting severe nausea. These adverse reactions generally occurred within the first 2 weeks of treatment and usually resolved within 4 weeks. The other most commonly reported adverse reactions included upper respiratory tract infections (8.4%), headache (7.9%), and tension headache (7.2%).
The most commonly reported adverse reactions in Phase 3 (Studies PALACE 1, PALACE 2, PALACE 3, PALACE 4 and ESTEEM 1 and ESTEEM 2) clinical studies have been gastrointestinal (GI) disorders including diarrhoea (15.7%) and nausea (13.9%). These GI adverse reactions were mostly mild to moderate in severity, with 0.3% of patients reporting severe diarrhoea and 0.3% of patients reporting severe nausea. These adverse reactions generally occurred within the first 2 weeks of treatment and usually resolved within 4 weeks. The other most commonly reported adverse reactions included upper respiratory tract infections (8.4%), headache (7.9%), and tension headache (7.2%).
 ACR 20 responses were higher in patients treated with Otezla than in patients treated with placebo when used alone or in combination with DMARDs. In Study PALACE 1, the proportion of patients with an ACR 20 response at Week 16 with concomitant DMARD use was 33.0% for Otezla 30 mg twice daily and 23.6% for placebo. The proportion of patients with an ACR 20 response at Week 16 without concomitant DMARD use was 46.8% for Otezla 30 mg twice daily and 10.3% for placebo. Similar results were observed in Studies PALACE 2 and PALACE 3.
ACR 20 responses were higher in patients treated with Otezla than in patients treated with placebo when used alone or in combination with DMARDs. In Study PALACE 1, the proportion of patients with an ACR 20 response at Week 16 with concomitant DMARD use was 33.0% for Otezla 30 mg twice daily and 23.6% for placebo. The proportion of patients with an ACR 20 response at Week 16 without concomitant DMARD use was 46.8% for Otezla 30 mg twice daily and 10.3% for placebo. Similar results were observed in Studies PALACE 2 and PALACE 3.
 Treatment with Otezla 30 mg twice daily resulted in greater improvement for each ACR component [number of swollen and tender joints, physician and patient assessment of disease activity and patient assessment of pain, HAQ-DI score and CRP value], compared to placebo at Weeks 16 and 24 in Study PALACE 4. Among patients who were continuously treated with Otezla, sustained improvements in individual ACR components were observed at Week 52.
Treatment with Otezla 30 mg twice daily resulted in greater improvement for each ACR component [number of swollen and tender joints, physician and patient assessment of disease activity and patient assessment of pain, HAQ-DI score and CRP value], compared to placebo at Weeks 16 and 24 in Study PALACE 4. Among patients who were continuously treated with Otezla, sustained improvements in individual ACR components were observed at Week 52. The clinical benefit of Otezla was demonstrated across multiple subgroups defined by baseline demographics, baseline clinical disease characteristics (including psoriasis disease duration and patients with a history of psoriatic arthritis), prior psoriasis medication usage and response to prior psoriasis treatments. Similar response rates were observed across all weight ranges.
The clinical benefit of Otezla was demonstrated across multiple subgroups defined by baseline demographics, baseline clinical disease characteristics (including psoriasis disease duration and patients with a history of psoriatic arthritis), prior psoriasis medication usage and response to prior psoriasis treatments. Similar response rates were observed across all weight ranges. Among patients originally randomised to Otezla, the sPGA, ScPGA, and Whole Body Itch responses were maintained through week 32.
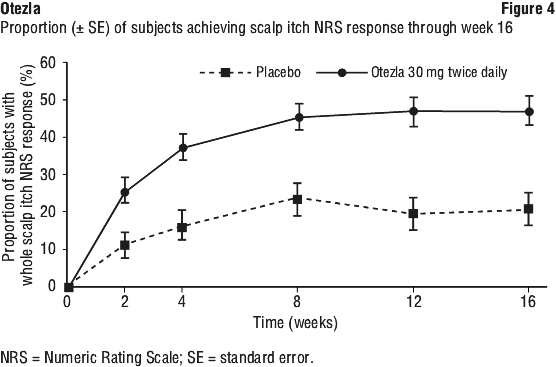
Among patients originally randomised to Otezla, the sPGA, ScPGA, and Whole Body Itch responses were maintained through week 32. Otezla 30 mg twice daily resulted in significant improvements in Whole Body Itch as demonstrated by the proportion of subjects with Whole Body Itch NRS response as early as Week 2 and at every visit through Week 16, compared with placebo (see Figure 3). Otezla 30 mg twice daily achieved similar improvement in Scalp Itch as demonstrated in Figure 4.
Otezla 30 mg twice daily resulted in significant improvements in Whole Body Itch as demonstrated by the proportion of subjects with Whole Body Itch NRS response as early as Week 2 and at every visit through Week 16, compared with placebo (see Figure 3). Otezla 30 mg twice daily achieved similar improvement in Scalp Itch as demonstrated in Figure 4.
 Among 104 patients originally randomised to Otezla 30 mg twice daily, 75 patients (approximately 72%) remained on this treatment at week 64. Among patients who were continuously treated with Otezla and remained in the study, improvements in oral ulcers and reduction of oral ulcer pain were maintained through week 64 (see Figure 5 and Figure 6).
Among 104 patients originally randomised to Otezla 30 mg twice daily, 75 patients (approximately 72%) remained on this treatment at week 64. Among patients who were continuously treated with Otezla and remained in the study, improvements in oral ulcers and reduction of oral ulcer pain were maintained through week 64 (see Figure 5 and Figure 6).