


What is in this leaflet
This leaflet answers some common questions about PALEXIA® SR.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you having PALEXIA® SR against the benefits they expect it will have for you.
If you have any concerns about this medicine, ask your doctor, nurse or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What PALEXIA® SR is used for
PALEXIA® (tapentadol) SR is used to relieve severe pain that is expected to be long lasting. This strong pain reliever belongs to a group of medicines known as opioid analgesics.
This medicine is available only with a doctor’s prescription.
PALEXIA® SR tablets are designed to release the pain reliever gradually over several hours.
Your doctor may have prescribed PALEXIA® SR for another reason.
Ask your doctor if you have any questions about why PALEXIA® SR has been prescribed for you.
Before you take PALEXIA® SR
When you must not take it
You must not take PALEXIA® SR if you:
- are allergic to tapentadol or any of the ingredients listed at the end of this leaflet. Signs of allergic reaction may include a skin rash, itching, shortness of breath or swelling of the face, lips or tongue
- have asthma or if your breathing is dangerously slow or shallow (respiratory depression, hypercapnia)
- have paralysis of the gut
- have acute poisoning with alcohol, sleeping pills, pain relievers or other psychotropic medicines (medicines that affect mood and emotions)
- are taking medicine for depression containing a monoamine oxidase inhibitor (MAOI) medicine (such as Nardil, Parnate) or have taken a MAOI within the last 14 days. Ask your doctor or pharmacist if any of your medicines is an MAOI.
Do not take PALEXIA® SR if the packaging is torn or shows signs of tampering or the tablets do not look quite right.
Do not take PALEXIA® SR if the expiry date on the pack has passed.
You should only start taking PALEXIA® SR under direct supervision of your doctor due to the following risks:
Addiction
You can become addicted to PALEXIA® SR even if you take it exactly as prescribed. PALEXIA® SR may become habit forming causing mental and physical dependence. If abused it may become less able to reduce pain.
Dependence
As with all other opioid containing products, your body may become used to you taking PALEXIA® SR. Taking it may result in physical dependence. Physical dependence means that you may experience withdrawal symptoms if you stop taking PALEXIA® SR suddenly, so it is important to take it exactly as directed by your doctor.
Tolerance
Tolerance to PALEXIA® SR may develop, which means that the effect of the medicine may decrease. If this happens, more may be needed to maintain the same effect.
Withdrawal
Continue taking your medicine for as long as your doctor tells you. If you stop having this medicine suddenly, your pain may worsen and you may experience some or all of the following withdrawal symptoms:
- nervousness, restlessness, agitation, trouble sleeping or anxiety
- body aches, weakness or stomach cramps
- loss of appetite, nausea, vomiting or diarrhoea
- increased heart rate, breathing rate or pupil size
- watery eyes, runny nose, chills or yawning
- increased sweating.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you are using any other medicines, including any that you buy without a prescription from a pharmacy, supermarket or health food shop. See also Taking other medicines.
Tell your doctor if you:
- have slow or shallow breathing
- have sleep related breathing disorders
- suffer from increased pressure in the brain or disturbed consciousness up to coma
- have had a head injury or a brain tumour
- have had an epileptic fit or seizure or if you have an increased risk of having epileptic fits or seizures
- suffer from a liver or kidney disease
- suffer from a pancreatic or biliary tract disease including pancreatitis
- are breastfeeding
- are pregnant, or planning to become pregnant
- have an addiction or history of abuse of alcohol, opioids or other drugs
- have been told that you have an intolerance to some sugars. Lactose is an ingredient in these tablets.
Tell your doctor if you are pregnant or planning to become pregnant. You should not take PALEXIA® SR during pregnancy unless your doctor has told you to do so. Taking PALEXIA® SR during pregnancy may lead to withdrawal symptoms in the newborn baby. You should not take PALEXIA® SR during labour as it can cause breathing problems and signs of withdrawal in the newborn.
Tell your doctor if you are breastfeeding. You should not take PALEXIA® SR if you are breastfeeding as it may pass into your breast milk. Your doctor will discuss the risks and benefits of using PALEXIA® SR.
Taking other medicines
Tell your doctor if you are using any other medicines, including any that you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines and PALEXIA® SR may interfere with each other. These medicines include:
- medicines for depression, sleeplessness or mental conditions such as selective serotonin reuptake inhibitors (SSRI’s), serotonin-norepinephrine reuptake inhibitors (SNRI’s), tricyclic anti-depressants (TCAs), monoamine oxidase inhibitor (MAOIs) and triptans
- other pain relievers such as morphine or codeine
- some cough medicines
- general anaesthetics such as propofol or midazolam (examples are Propofol™ or Midazolam™)
- medicines that slow the brain activity (central nervous system (CNS) depressants or phenothiazines). These medicines can be used to treat anxiety, muscle tension, pain, insomnia, acute stress reactions, panic attacks or seizure disorders such as sleeping pills, tranquilizers, hypnotics or sedatives (examples are Stelazine™, Largactil™, Valium™, Temaze™, or Xanax™).
- a group of medicines called anticholinergics. These medicines can be used to treat a wide range of medical conditions including asthma, a respiratory condition called chronic obstructive pulmonary disorder (COPD), Parkinson's Disease, cardiovascular disease, urinary incontinence (loss of bladder control), psychiatric disorders including schizophrenia, anxiety, depression, allergies and travel sickness/nausea.
Taking these medicines with PALEXIA® SR may increase the risk of possible side effects (see Side Effects). Your breathing may become seriously slow or shallow (respiratory depression) and your blood pressure may decrease. Your consciousness may be decreased, you may feel drowsier or feel that you might faint. If this happens tell your doctor.
Do not take PALEXIA® SR with alcohol. Some side effects such as drowsiness may be increased.
Other medications can also interfere with PALEXIA® SR and make you feel drowsy (see Warning information at the start of this document). Ask your doctor or pharmacist for more information.
How to take PALEXIA® SR
You should only start taking PALEXIA® SR under the direct supervision of your doctor.
Your doctor will tell you how much you should take, when and how often. It is important that you take this medicine as directed by your doctor.
If you are unsure, ask your doctor or pharmacist.
How much to take
The usual dose is 1 tablet every 12 hours.
Your doctor may prescribe a different, more appropriate dose or interval of dosing, if this is necessary for you.
If you feel that the effect of these tablets is too strong or too weak, talk to your doctor or pharmacist.
PALEXIA® SR is not suitable for children and adolescents below the age of 18 years.
In elderly patients (above 65 years) usually no dose adjustment is necessary. However, the excretion of tapentadol may be delayed in some patients of this age group. If this applies to you, your doctor may recommend a different dosage regimen.
Patients with severe liver problems should not take these tablets. If you have moderate problems, your doctor will recommend a different dosage regimen.
Patients with severe kidney problems should not take these tablets.
Carefully follow all directions given to you by your doctor and pharmacist. These directions may differ from the information in this leaflet.
When and how should you take the tablets
PALEXIA® SR tablets should be swallowed whole with water. They may be taken, before, with, or after food.
PALEXIA® SR tablets are only designed to work properly if swallowed whole. The tablets may release all their contents at once if broken, chewed, crushed or dissolved, which can be dangerous and cause serious problems, such as an overdose which may be fatal. Unless stated otherwise, they should be swallowed whole.
The shell of the PALEXIA® SR tablets may appear in your stool. This is not a cause for concern as your body will have already absorbed the active ingredient.
How long to take it
This differs between individuals depending on how severe your pain is, how you respond to PALEXIA® SR and the cause of your pain. Ask your doctor for advice on how long you need to take PALEXIA® SR.
Carefully follow all directions given to you by your doctor and pharmacist. These directions may differ from the information contained in this leaflet.
If you forget to take it
If you forget to take a dose, you can take it as soon as you remember. The next dose should be taken after twelve hours, or as prescribed by your doctor.
Do not take a double dose to make up for the dose that you missed.
If you take too much (overdose)
If you or someone else receive too much (overdose), and experience one or more of the symptoms below, call triple zero (000) for an ambulance. Keep the person awake by talking to them or gently shaking them every now and then. You should follow the above steps even if someone other than you have accidentally used PALEXIA® SR that was prescribed for you. If someone takes an overdose, they may experience one or more of the following symptoms:
- Slow, unusual or difficult breathing
- Drowsiness, dizziness or unconsciousness
- Slow or weak heartbeat
- Nausea or vomiting
- Convulsions or fits
If you think you or someone else may have taken too much PALEXIA® SR, you should immediately:
- phone the Poisons Information Centre (by calling 13 11 26), or
- contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
When seeking medical attention, take this leaflet and remaining medicine with you to show the doctor. Also tell them about any other medicines or alcohol which have been taken.
While you are taking PALEXIA® SR
Things you must do
Be sure to keep all of your doctor’s appointments so that your progress can be checked.
If you are about to be started on any new medicine, tell your doctor and pharmacist that you are taking PALEXIA® SR.
Tell your doctor if you believe that PALEXIA® SR is not helping your condition.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed. Otherwise your doctor may think it is not working effectively and change your treatment unnecessarily.
If you become pregnant while you are taking PALEXIA® SR, tell your doctor immediately.
If you plan to have surgery, even at the dentist’s, tell your doctor, anaesthetist or dentist that you are taking this medicine. It may affect other medicines used during surgery.
Things you must not do
If you wish to stop treatment, please tell your doctor first before stopping treatment. Do not stop taking this medicine unless your doctor tells you to. If your doctor wants you to stop taking your tablets, he/she will tell you how to do this. This may include a gradual reduction in the dose.
Some people may feel unwell if they suddenly stop taking PALEXIA® SR. If you suddenly stop taking PALEXIA® SR you may experience withdrawal symptoms (see Withdrawal section above).
Do not give PALEXIA® SR to anyone else, even if they have the same condition as you.
Do not take PALEXIA® SR for any other complaints unless your doctor tells you to.
Things to be careful of
Do not drive or operate heavy machinery until you know how PALEXIA® SR affects you.
PALEXIA® SR can make you sleepy, dizzy, or lightheaded.
Do not drink alcohol while you are taking PALEXIA® SR tablets. Drinking alcohol while taking PALEXIA® SR may make you feel more sleepy and increase the risk of serious side effects, such as shallow breathing, with the risk of stopping breathing and loss of consciousness.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking PALEXIA® SR.
Like all medicines, PALEXIA® SR can cause some unwanted side effects in some people. Sometimes they are serious, most of the time they are not. Side effects not listed in this leaflet may occur in some patients. You may need to get medical attention if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Very common:
- nausea
- constipation
- dizziness
- drowsiness
- headache.
Common:
- decreased appetite
- anxiety
- depressed mood
- difficulty sleeping
- nervousness
- restlessness
- inability to concentrate
- trembling
- muscle twitches
- flushing or reddening of the face and neck
- shortness of breath
- vomiting
- diarrhoea
- indigestion
- itching
- increased sweating
- rash
- feeling of weakness
- fatigue
- feeling of body temperature change
- mucosal dryness
- accumulation or retention of water in the tissues (oedema).
Uncommon:
- allergic reaction
- weight loss
- disorientation
- confusion
- excitability (agitation)
- perception disturbances
- abnormal dreams
- euphoric mood
- depressed level of consciousness
- memory impairment
- mental impairment
- fainting
- sedation
- balance disorder
- difficulty speaking
- numbness
- abnormal sensations of the skin (e.g. tingling, prickling)
- abnormal vision
- faster or irregular heart beat
- slower heart beat
- decreased blood pressure
- abdominal discomfort
- hives
- delay in passing urine
- frequent urination
- sexual dysfunction
- drug withdrawal syndrome (see “Things you must not do”)
- feeling abnormal
- irritability
Rare:
- drug dependence
- thinking abnormal thoughts
- epileptic fit
- near fainting
- abnormal coordination
- dangerously slow or shallow breathing
- impaired gastric emptying
- feeling drunk
- feeling of relaxation.
If any of the following happen, tell your doctor or go to accident and emergency at your nearest hospital immediately:
- skin rash (red spots or patches), itching, hives
- swelling of the face, lips, mouth or throat which may cause difficulty in swallowing or breathing
- chest tightness, wheezing or pain in the chest
- heart palpitations
- faintness or collapse
- hallucinations
- seizures, fits or convulsions.
These are very serious side effects. If you have them, you may have had a serious allergic reaction to PALEXIA® SR. You may need urgent medical attention or hospitalisation.
If you notice any unwanted effects not mentioned in this leaflet, please inform your doctor, or pharmacist.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using PALEXIA® SR
Storage
Keep PALEXIA® SR tablets in the blister pack until it is time to take them.
Keep your tablets in a cool, dry place where the temperature stays below 30°C.
Do not store PALEXIA® SR in the bathroom or near a sink.
Do not leave them in a car or on a window sill Heat and dampness can destroy some medicines.
Keep PALEXIA® SR tablets where children cannot reach them. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If the medicine is damaged, you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
Product Description
There are 6 distinct strengths of PALEXIA® SR sustained release tablets:
PALEXIA® SR 25 mg tablets
Slightly brownish-orange film-coated oblong shaped tablets (5.5 mm x 10 mm) marked with Grünenthal logo on one side and “H9” on the other side.
PALEXIA® SR 50 mg tablets
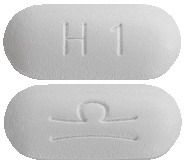
White film-coated oblong shaped tablets (6.5 mm x 15 mm) marked with Grünenthal logo on one side and “H1” on the other side.
PALEXIA® SR 100 mg tablets
Pale yellow film-coated oblong shaped tablets (6.5 mm x 15 mm) marked with Grünenthal logo on one side and “H2” on the other side.
PALEXIA® SR 150 mg tablets
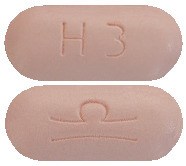
Pale pink film-coated oblong shaped tablets (6.5 mm x 15 mm) marked with Grünenthal logo on one side and “H3” on the other side.
PALEXIA® SR 200 mg tablets
Pale orange film-coated oblong shaped tablets (7 mm x 17 mm) marked with Grünenthal logo on one side and “H4” on the other side.
PALEXIA® SR 250 mg tablets
Brownish red film-coated oblong shaped tablets (7 mm x 17 mm) marked with Grünenthal logo on one side and “H5” on the other side.
The registered pack sizes for PALEXIA® SR tablets are 7, 10, 14, 20, 28, 30, 40, 50, 56, 60, 90 and 100 tablets.
All strengths and pack sizes may not be available.
PALEXIA® SR tablets are sealed in a blister pack.
Ingredients
Active ingredients
PALEXIA® SR 25 mg tablets - each tablet contains 25 mg tapentadol (as hydrochloride).
PALEXIA® SR 50 mg tablets - each tablet contains 50 mg tapentadol (as hydrochloride).
PALEXIA® SR 100 mg tablets - each tablet contains 100 mg tapentadol (as hydrochloride).
PALEXIA® SR 150 mg tablets - each tablet contains 150 mg tapentadol (as hydrochloride).
PALEXIA® SR 200 mg tablets - each tablet contains 200 mg tapentadol (as hydrochloride).
PALEXIA® SR 250 mg tablets - each tablet contains 250 mg tapentadol (as hydrochloride).
Inactive ingredients
PALEXIA® SR tablets also contain the following inactive ingredients:
- hypromellose 100,000 mPa-s
- microcrystalline cellulose
- colloidal anhydrous silica
- magnesium stearate
- hypromellose 6 mPa-s
- lactose monohydrate
- purified talc
- macrogol 6000
- titanium dioxide (E171)
- propylene glycol (50, 100, 150, 200 and 250 mg tablets only)
- macrogol 400 (25 mg tablets only)
- iron oxide yellow (E172) (25, 100, 150, 200 and 250 mg tablets only)
- iron oxide red (E172) (25, 150, 200 and 250 mg tablets only)
- iron oxide black (E172) (250 mg tablets only).
PALEXIA® SR contains lactose.
PALEXIA® SR does not contain:
- gluten
- preservative.
Further information
You can obtain more information from your doctor or pharmacist.
Australian sponsor:
Seqirus Pty Ltd
ABN 26 160 735 035
655 Elizabeth Street
Melbourne VIC 3000
Australia
www.cslseqirus.com.au
PALEXIA® SR is distributed in Australia by:
Seqirus (Australia) Pty Ltd
ABN 66 120 398 067
63 Poplar Road
Parkville, VIC 3052
Australia
PALEXIA® SR is manufactured by:
Grünenthal GmBH, Germany
Australian Registration Numbers:
PALEXIA® SR 25 mg tablets
AUST R 229737
PALEXIA® SR 50 mg tablets
AUST R 165332
PALEXIA® SR 100 mg tablets
AUST R 165346
PALEXIA® SR 150 mg tablets
AUST R 165347
PALEXIA® SR 200 mg tablets
AUST R 165356
PALEXIA® SR 250 mg tablets
AUST R 165357
This leaflet was prepared in February 2025.
PALEXIA® is a registered trademark of Grünenthal GmBH, used under licence.
Published by MIMS April 2025


 The following adverse drug reactions (ADRs) were reported from clinical trials performed with Palexia SR.
The following adverse drug reactions (ADRs) were reported from clinical trials performed with Palexia SR.



