What is in this leaflet
This leaflet answers some common questions about this medicine. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may want to read it again.
The information in this leaflet was last updated on the date listed on the last page. More recent information on this medicine may be available
What this medicine is used for
The name of your medicine is Pelgraz. It contains the active ingredient, pegfilgrastim.
Pelgraz is used to help fight infection, following chemotherapy. It belongs to a group of medicines called immunostimulants.
Some chemotherapy will reduce the number of neutrophils in your body. Although Pelgraz is not a treatment for cancer, it does help the body to make new neutrophils. This will reduce your chance of developing infections that might require antibiotics and/or hospital stays. It may even increase your chance of receiving your chemotherapy on time and at the right dose.
How it works
Pelgraz is a long acting form of Recombinant Human Granulocyte Colony Stimulating Factor or G-CSF.
G-CSF is produced in the bone marrow and assists in the production of neutrophils, which are a type of white blood cell. Neutrophils help the body fight infections by surrounding and destroying the bacteria that cause the infections. G-CSF also helps neutrophils to do this work better.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
Before you use or are given this medicine
When you must not use it
You must not use or be given this medicine if you have an allergy to:
- any medicine containing pegfilgrastim or filgrastim
- any medicines or products that are produced using the bacteria, E.coli
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue, throat or other parts of the body
- rash, itching or hives on the skin.
Do not use Pelgraz at the same time as your chemotherapy or radiotherapy.
Do not use Pelgraz within 24 hours after you receive chemotherapy. This is because the chemotherapy medicine may stop Pelgraz from increasing the number of infection-fighting neutrophils.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should use or be given this medicine, talk to your doctor.
Before you use or are given it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- a medical condition affecting the bone marrow or blood
- a family history of a genetic disorder
- sickle cell disease
- problems with your kidneys, liver, heart or other organs
- previous treatment for cancer
- any infections, cancers or tumours.
Tell your doctor if you are pregnant or plan to become pregnant, or are breastfeeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor any of the above, tell him/her before you use or are given this medicine.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
These medicines may be affected by this medicine or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How this medicine is used or given
This medicine is given by injection, into the tissues, just below the skin. This is called a subcutaneous injection.
Your doctor, nurse or pharmacist may suggest that you or your carer be taught how to give a subcutaneous injection. This will allow you to give yourself the injection at home.
It is important that you get special training from your doctor or nurse before you give yourself the injection.
If you are not sure about giving yourself the injection or you have any questions, ask your doctor, nurse or pharmacist for help.
Carefully follow all directions given to you by your doctor, nurse or pharmacist. They may differ from the information in this leaflet.
If you do not understand the instructions in this leaflet, ask your doctor, pharmacist or nurse for help.
How much to inject
The usual dose is one subcutaneous injection (6 mg/0.6 mL), 24 hours after the end of each chemotherapy cycle.
How to give yourself the injection
This section contains information on how to give yourself an injection of Pelgraz (pre-filled syringe with passive needle guard).
Before you give yourself the injection, read this important information:
- Tell your doctor if you have an allergy to latex. The needle cover on the pre-filled syringe contains a derivative of latex and may cause severe allergic reactions.
- DO NOT remove the needle cover from the pre-filled syringe until you are ready to inject.
- DO NOT use the pre-filled syringe if it has been dropped on a hard surface. Use a new pre-filled syringe and call your doctor or healthcare provider.
- DO NOT attempt to remove the clear pre-filled syringe needle guard from the pre-filled syringe.

Things to do before you inject
Remove the pre-filled syringe tray from the package and gather the supplies needed for your injection: alcohol wipes, a cotton ball or gauze pad, an adhesive bandage and a sharps disposal container (may be provided).
For a more comfortable injection, leave the pre-filled syringe at room temperature for about 30 minutes before injecting.
On a clean, well-lit work surface, place the new pre-filled syringe and the other supplies.
- DO NOT try to warm the syringe by using a heat source, such as hot water or microwave.
- DO NOT leave the pre-filled syringe exposed to direct sunlight.
- DO NOT shake the pre-filled syringe.
- Keep pre-filled syringes out of the sight and reach of children.
Follow these below instructions for correct handling technique when removing the pre-filled syringe with the passive needle guard from the packaging; otherwise, the needle’s safety mechanism may be triggered, making the syringe unusable.
1. Open the tray, peeling away the cover.
To remove the individual syringe from the blister packaging, start with the corner (as shown by the arrow and “Peel here”) and open the blister pack by completely peeling back the top layer

2. Hold onto the pre-filled syringe safety guard to remove the prefilled syringe from the tray.
Remove the syringe from the blister pack by the body as shown below:
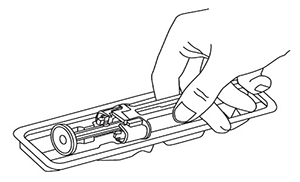
For safety reasons:
- DO NOT lift the product by the plunger or needle cover.
- DO NOT touch the needle guard activation clips at any time during use. This may trigger the needle's safety mechanism causing the needle to retract (pull back) before your injection is given. This will make the syringe unusable.
3. Inspect the medicine and prefilled syringe. DO NOT use the pre-filled syringe if:
- the medicine is cloudy or there are particles in it. It must be a clear and colourless liquid
- any part appears cracked or broken
- the needle cover is missing or not securely attached
- the expiry date printed on the label has passed the last day of the month shown
- in all cases, call your doctor or healthcare provider.
Selecting and preparing the injection site
You will need to give yourself an injection into the tissue under the skin, known as a subcutaneous injection:
- Wash hands thoroughly with soap and water.
- Before you inject this medicine, you must always disinfect the skin on the selected injection site by using an alcohol swab. Let the skin dry.
- Choose an injection site from the most suitable place on your body (listed and shown here):
- outer area of your upper arms
- front of your middle thighs
- abdomen, except for the 5 centimetre area around the navel
- upper outer area of your buttocks

- DO NOT touch the selected injection area before injecting.
- DO NOT inject into areas where the skin is tender, bruised, red, or hard. Avoid injecting into areas with scars or stretch marks.
- From the above options, change the injection site each time you take an injection so that you do not develop soreness in one area.
Injecting the prescribed dose of medicine
- Before you inject this medicine, you must always clean the skin on the selected injection site by using an alcohol wipe.
- Hold the pre-filled syringe by the body (needle guard) with the needle pointing up and away from your body, as this helps to prevent the medicine from accidentally expelling from the syringe.
- DO NOT remove the needle cover from the pre-filled syringe until you are ready to inject.
- Carefully remove the needle cover straight off, without twisting it.

- DO NOT touch the needle or the plunger.
- DO NOT use if the syringe is damaged or the needle is bent.
- DO NOT use the pre-filled syringe if it has been dropped on a hard surface. The prefilled syringe may be broken even if you cannot see the break. Use a new pre-filled syringe.
- DO NOT push the plunger on the syringe before injection. This may release the needle guard by springing and completely covering the needle.
- DO NOT use the pre-filled syringe if any liquid is accidentally expelled from the syringe. Dispose of that syringe in a puncture-proof disposal container. Use a new pre-filled syringe.
- DO NOT push the plunger on the syringe before injection. This may release the needle guard by springing and completely covering the needle.
- Hold the pre-filled syringe between the thumb and the forefinger of the hand that you will use to inject the medicine.
- Use the other hand to pinch a fold of the skin at the cleaned injection site between your thumb and forefinger without squeezing it, as shown below:

- Insert the needle into the skin at an angle greater than or equal to 45° (45 degrees), as shown by your nurse, doctor or pharmacist, and as shown below:

- After the needle is inserted, let go pinching on the fold of skin.
Inject the prescribed dose subcutaneously by pushing the plunger with your thumb while grasping the finger grips, as directed by your doctor, nurse, or pharmacist, and as shown below:

- Press the plunger slowly and completely, until all of the medicine has been injected, as shown below:

The needle guard will not be activated unless the entire dose has been administered and you remove downward pressure on the plunger.
- When the syringe is emptied of all the medicine, slowly lift your thumb from the plunger, which will release the needle guard. The needle will then withdraw from the skin and be covered and locked in place by the needle guard.

- After the injection, immediately place cotton or gauze on the injected site and apply pressure for several seconds.
Disposal
Place the pre-filled syringe with the needle guard-covered needle into a puncture-proof container for proper disposal.
Medicines should be disposed of in accordance with local requirements. These measures will help protect the environment.
Follow the instructions given by your doctor, nurse, or pharmacist on how to properly dispose of containers with used syringes and needle guards.
Always keep the disposal container out of the reach and sight of children.
- DO NOT re-use the pre-filled syringes.
- DO NOT recycle pre-filled syringes.
- DO NOT throw the pre-filled syringes into household bins.
Examining the injection site
If there is blood, press a cotton ball or gauze pad on your injection site. DO NOT rub the injection site.
Apply an adhesive bandage if needed.
If you have any problems, please ask your doctor, nurse or pharmacist for help and advice.
When to inject
Use this medicine 24 hours after the end of each chemotherapy cycle.
Your doctor will tell you when to begin your treatment and when to stop.
If you forget your injection
If you miss your scheduled dose, talk to your doctor, nurse or pharmacist as soon as possible.
If you receive too much (overdose)
If you inject more of this medicine than you need, talk to your doctor, nurse or pharmacist. If you feel unwell in any way you talk to your doctor, nurse or pharmacist immediately.
While you are using this medicine
Things you must do
Watch for any signs or symptoms of infection.
There are many ways an infection may show itself.
Symptoms of an infection include:
- fever (a temperature of 38.2°C or greater, or as your doctor suggests)
- chills
- rash
- sore throat
- diarrhoea
- earache
- difficult or painful breathing, coughing or wheezing.
Go straight to your hospital if you develop any of these symptoms.
If you are about to be started on any new medicine, remind your doctor, nurse and pharmacist that you are using this medicine.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your health can be monitored. Your doctor may order blood tests to check the levels of infection-fighting neutrophils and other blood cells, as well as to investigate unwanted side effects.
Things you must not do
Do not use this medicine to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Side effects
Tell your doctor as soon as possible if you do not feel well after using this medicine or if you have any questions or concerns.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
If you are over 65 years of age, you may have an increased chance of getting side effects.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor, nurse or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- temporary bone pain, such as in the lower back or in the long bones of the arms or legs.
This pain is usually relieved with non-prescription painkillers, such as paracetamol. If you continue to have bone pain even after having taken this form of pain relief, you should speak to your doctor, as you may need a prescription medicine. - headache
- general aches and pains in joints and muscles
- reddish or purplish blotches under the skin
- injection site pain and redness of the skin at the injection site.
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- pain in the upper left side of the stomach (abdomen)
- left shoulder pain
- dizziness
- fever and painful skin lesions most commonly on your arms, legs and sometimes on your face and neck
- blood in the urine.
The above list includes serious side effects that may require medical attention.
If any of the following happen, tell your doctor or nurse immediately, or go to the Emergency department at your nearest hospital:
- swelling or puffiness
- less frequent urination
- swelling of your stomach-area (abdomen) and feeling of fullness
- general feeling of tiredness.
These may be serious side effects. You may need urgent medical attention.
If any of the following happen, stop taking this medicine and go to the Emergency department at your nearest hospital, as you may need urgent medical attention:
- rash over a large area of the body, itching or hives
- shortness of breath, wheezing or difficulty breathing
- coughing up blood, bleeding from the lung
- swelling of the face, lips, tongue or other parts of the body
- faintness
- rapid pulse or sweating.
The above list includes very serious side effects. If you have them you may have had a serious allergic reaction to this medicine. You may need urgent medical attention or hospitalisation.
Tell your doctor, nurse or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may occur in some people.
Storage and presentation
Storage
Keep this medicine in a refrigerator at a temperature of 2°C to 8°C.
This medicine can be removed from the refrigerator and left at room temperature (not above 25°C) for a single period of up to 15 days that ends within the labelled expiry date. Once it has been out at room temperature, it should not be put back into the refrigerator.
You should avoid freezing this medicine; however, if it has been accidentally frozen, allow the medicine to thaw in the refrigerator before use.
Do not use this medicine if it has been frozen a second time.
Keep your medicine in the carton. Protect it from light.
Keep it where children cannot reach it.
Disposal
Once you have finished the injection, do not put the needle cover back on the used syringe.
Discard the used syringe into an approved, puncture-resistant sharps container, as instructed by your doctor, nurse or pharmacist. Keep it out of the reach of children.
Never put the used syringes into your normal household rubbish bin.
Use each pre-filled syringe only for one injection.
DO NOT recycle.
DO NOT try to put the needle cover back on the needle.
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Pelgraz is a clear, colourless solution. It is supplied in a carton as a pre-filled syringe with a passive needle guard.
Ingredients
Each syringe contains 6 mg of pegfilgrastim, as the active ingredient.
This medicine also contains the following:
- 0.35 mg glacial acetic acid
- 30 mg sorbitol
- 0.024 mg polysorbate 20
- 0.034 mg sodium hydroxide
- water for injection (to 0.6 mL).
This medicine does not contain gluten, lactose, sucrose, tartrazine or other azo dyes.
Australian Registration Number
Pelgraz 6 mg/0.6 mL solution for injection, pre-filled syringe: AUST R 308177.
Supplier
Accord Healthcare Pty Ltd
Level 24, 570 Bourke Street
Melbourne, VIC, 3000
Australia
This leaflet was prepared in November 2019.
Published by MIMS September 2020

 Across all studies, no life-threatening or fatal adverse events were attributed to pegfilgrastim. In these studies, there was only 1 serious adverse event (dyspnoea) reported in a single patient as possibly related to pegfilgrastim.
Across all studies, no life-threatening or fatal adverse events were attributed to pegfilgrastim. In these studies, there was only 1 serious adverse event (dyspnoea) reported in a single patient as possibly related to pegfilgrastim.

 In study 2, patients with breast cancer (n = 301) were randomised to receive a single injection of pegfilgrastim 100 microgram/kg or daily injections of filgrastim 5 microgram/kg/day after each of 4 cycles of the highly myelosuppressive chemotherapy regimen doxorubicin and docetaxel (AT). In cycle 1, a single SC injection of pegfilgrastim resulted in a duration of severe neutropenia that was clinically and statistically similar to that observed after a mean of 11 daily injections of filgrastim (see Table 5). Durations of severe neutropenia were also comparable between treatment groups in all subsequent cycles. There is a significant difference in the incidence of febrile neutropenia between the groups in Study 2.
In study 2, patients with breast cancer (n = 301) were randomised to receive a single injection of pegfilgrastim 100 microgram/kg or daily injections of filgrastim 5 microgram/kg/day after each of 4 cycles of the highly myelosuppressive chemotherapy regimen doxorubicin and docetaxel (AT). In cycle 1, a single SC injection of pegfilgrastim resulted in a duration of severe neutropenia that was clinically and statistically similar to that observed after a mean of 11 daily injections of filgrastim (see Table 5). Durations of severe neutropenia were also comparable between treatment groups in all subsequent cycles. There is a significant difference in the incidence of febrile neutropenia between the groups in Study 2.
 The results of DSN data in Cycle 1 show the similarity in efficacy of Pelgraz and US-Neulasta (-0.04 to 0.50) as well as of Pelgraz and EU-Neulasta (-0.29 to 0.26) since the 95% CI are within the equivalence range of -0.5 to 0.5 days for both comparisons.
The results of DSN data in Cycle 1 show the similarity in efficacy of Pelgraz and US-Neulasta (-0.04 to 0.50) as well as of Pelgraz and EU-Neulasta (-0.29 to 0.26) since the 95% CI are within the equivalence range of -0.5 to 0.5 days for both comparisons.
 Table 9 shows results from the potency corrected pegfilgrastim concentration data for both Pelgraz and US-Neulasta. For the primary pharmacokinetic endpoint of AUCt, the 90% confidence interval of the Pelgraz/Neulasta ratio of geometric means was contained within the acceptance range of 80 - 125%.
Table 9 shows results from the potency corrected pegfilgrastim concentration data for both Pelgraz and US-Neulasta. For the primary pharmacokinetic endpoint of AUCt, the 90% confidence interval of the Pelgraz/Neulasta ratio of geometric means was contained within the acceptance range of 80 - 125%.

