1 Name of Medicine
Daunorubicin (as hydrochloride).
2 Qualitative and Quantitative Composition
Each vial of Pfizer Daunorubicin Powder for Injection contains 20 mg of daunorubicin hydrochloride (equivalent to 18.7 mg of daunorubicin).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Powder for injection.
Pfizer Daunorubicin Powder for Injection is a red-orange powder in a clear glass vial to be reconstituted for intravenous use.
4.1 Therapeutic Indications
Pfizer Daunorubicin Powder for Injection is indicated for the treatment of the following:
Acute lymphocytic (lymphoblastic) leukaemia.
Daunorubicin is usually reserved for use in cases shown to be resistant to other drugs. However, combined treatment with daunorubicin, vincristine and a steroid has been used in the early stages of this disease.
Acute myeloblastic leukaemia.
Daunorubicin has been used in all stages, alone or in combination with other cytotoxic agents (e.g. cytarabine).
Disseminated solid tumours.
Daunorubicin has been investigated for use in these tumours and found to be effective in some cases of disseminated neuroblastoma and rhabdomyosarcoma.4.2 Dose and Method of Administration
Dosage.
The dosage of each individual injection may vary from 0.5 to 3 mg/kg, with the frequency of repetition according to the dose:
0.5 to 1 mg/kg repeated at intervals of one or more days;
2 mg/kg repeated at intervals of four or more days;
2.5 or 3 mg/kg, if used, should only be given at seven to fourteen day intervals.
Dosage must be adjusted to meet individual requirements of each patient, on the basis of clinical response and appearance or severity of toxicity. One injection has sometimes sufficed; commonly three to six injections have been necessary; occasionally up to 10 injections in one series have been used.
When second or subsequent injections are to be given the doses and the time intervals depend on the effect of the previous doses and must be the subject of careful deliberation, examination of the peripheral blood and under some circumstances, of the marrow.
The dosage of daunorubicin is usually based on the patient's body surface area (m2), but in paediatric patients younger than 2 years of age (or with a body surface area of less than 0.5 m2) it is suggested to calculate the dosage on the body weight (kg) rather than on body surface area.
For paediatric patients under 2 years of age (or below 0.5 m2 body surface area), the maximum cumulative dose is 10 mg/kg.
For paediatric patients over 2 years, the maximal cumulative dose is 300 mg/m2.
For information on cardiac induced toxicity and cardiomyopathy in paediatric patients, see Section 4.4 Special Warnings and Precautions for Use, Paediatric use.
Acute lymphocytic leukaemia. Doses of 1 mg/kg may be repeated according to tolerance and effect at one to four day intervals.
Acute myeloblastic leukaemia. Each dose should be about 2 mg/kg, more or less, according to effect, repeated at four to seven day intervals. Doses of over 2 mg/kg should be employed with caution at intervals of one week or longer.
Used in combination with other cytotoxic. When daunorubicin is administered with other cytotoxic drugs, which have a tendency to depress the marrow, the dosage should be suitably reduced. Examples of combination dosage regimens are as follows:
Acute lymphocytic leukaemia.
Prednisolone 100 mg/m2 daily with vincristine 1.5 mg/m2 on the first and second days of each week, until remission occurs.
Acute myeloblastic leukaemia.
Repeated intermittent courses of daunorubicin and cytosine arabinoside, each course consisting of the intravenous injection of both daunorubicin 1.5 mg/kg and cytarabine 2 mg/kg on the first day, followed by daily cytarabine only for a further four days. It has been reported that an average of two to three such courses at ten day intervals is required to induce remission.
Due to cardiotoxicity, the total lifetime dosage should not exceed 20 mg/kg (see Section 4.4 Special Warnings and Precautions for Use, Cardiac toxicity). The drug is therefore not suitable for maintenance therapy.
Method of administration.
Pfizer Daunorubicin Powder for Injection, following reconstitution, is intended for intravenous use only. The reconstituted solution should not be administered by either the intramuscular or the subcutaneous routes, as severe tissue necrosis will result.
Daunorubicin should be administered only under the supervision of a physician who is experienced in the use of cancer chemotherapeutic agents.
Reconstitution/preparation for administration.
Prior to use, the contents of each vial should be reconstituted with 9.3 mL of 0.9% sodium chloride solution. The daunorubicin concentration of the reconstituted solution is 2 mg/mL.
Following reconstitution, it is recommended the reconstituted solution be added to a free flowing IV infusion of 0.9% sodium chloride or 5% glucose injection. The tubing should be connected to a butterfly needle, inserted preferably into a large vein. The dose and the size of the vein will determine the rate of administration, which should not be less than 3-5 minutes. Erythematous streaking and facial flushing are indications of too rapid administration.
A direct push injection is not recommended due to the risk of extravasation, which may occur even in the presence of adequate blood return upon needle aspiration.
Product is for single use in one patient only. Discard any residue.
Instructions to be given to patients.
Patients should be advised to immediately report any stinging or burning as these indicate possible extravasations. If this occurs the infusion should be stopped, removed and restarted in another vein.
Daunorubicin may transiently impart a red discolouration to the urine after administration; patients should be advised to expect this.
Complete alopecia is common but reversible on withdrawal of treatment. Patients should be made aware of this adverse effect before commencement of treatment.
Due to the increased frequency of infection as well as haemorrhagic complications resulting from daunorubicin therapy, the patient should be instructed to notify the doctor if fever, sore throat, or unusual bleeding or bruising occurs.
Dosage adjustment.
Hepatic impairment.
Daunorubicin should not be administered to patients with severe hepatic impairment (Child-Pugh Grade C [total score 10-15]) (see Section 4.3 Contraindications).
For patients with mild and moderate hepatic impairment (Child-Pugh Grade A [total score 5-6] and B [total score 7-9]), dose reductions are recommended based on the following serum bilirubin values:
Bilirubin 1.2 to 3 mg/dL: one-half of recommended starting dose.
Bilirubin > 3 mg/dL: one-fourth of recommended starting dose.
Renal impairment.
If serum creatinine is above 3.0 mg/dL, the daunorubicin dose should be reduced by one-half.4.3 Contraindications
Pfizer Daunorubicin Powder for Injection is contraindicated in patients:
with persistent myelosuppression; or marked myelosuppression induced by previous treatment with other cytotoxic agents or radiotherapy;
with impaired cardiac function (including myocardial insufficiency, recent myocardial infarction and severe arrhythmias);
who have previously received the full cumulative dose of daunorubicin and/or doxorubicin and/or other anthracyclines and/or anthracenediones (see Section 4.4 Special Warnings and Precautions for Use, Cardiac toxicity);
with known hypersensitivity to daunorubicin or any other component of the product, other anthracyclines, or anthracenediones;
with severe infections;
with severe hepatic (Child-Pugh Grade C [total score 10-15]) or renal function impairment (GFR < 10 mL/min or serum creatinine > 7.9 mg/dL);
who are pregnant. See Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy.
4.4 Special Warnings and Precautions for Use
Daunorubicin should be administered only under the supervision of a physician who is experienced in the use of cancer chemotherapeutic agents.
Initial treatment with daunorubicin requires close observation of the patient and extensive laboratory monitoring. It is recommended, therefore, that patients be hospitalised at least during the first phase of treatment. Blood counts and monitoring of parameters of renal and liver function should be performed prior to each treatment with daunorubicin.
Administration of myelosuppressive drugs such as daunorubicin may lead to an increased frequency of infections and haemorrhagic complications. These complications are potentially fatal therefore patients should be instructed to notify the physician if fever, sore throat, or unusual bruising or bleeding occurs.
Patients should recover from acute toxicities of prior cytotoxic treatment (such as stomatitis, neutropenia, thrombocytopenia, and generalised infections) before beginning treatment with daunorubicin.
Cardiac toxicity.
Special attention must be given (by close cardiac monitoring) to the cardiac toxicity exhibited by daunorubicin, especially in infants and children.
Daunorubicin has a cardiotoxic effect, which can be manifested under two distinct sets of circumstances:
Firstly, daily administration of large doses of ≥ 2 mg/kg (or ≥ 55 mg/m2) will result in transient reversible electrocardiogram (ECG) changes in a proportion of cases. This can be avoided by administering the drug at longer intervals.
Secondly, exceeding the total cumulative dose of 20 mg/kg (or 550 mg/m2) may result in irreversible cardiac failure. This can occur with very little warning and after only a short period of tachycardia.
Cardiomyopathy usually appears within 1 to 6 months after initiation of therapy. It may develop suddenly and may not be detected by routine ECG. It may be irreversible and fatal but responds to treatment if detected early.
Acute and delayed cardiotoxicity. Cardiotoxicity may be manifested by early (i.e. acute) or late (i.e. delayed) events.
Early (i.e. acute) events.
Early cardiotoxicity of daunorubicin consists mainly of sinus tachycardia and/or ECG abnormalities such as non-specific ST-T wave changes. Tachyarrhythmias, including premature ventricular contractions, as well as heart block have also been reported. These effects do not usually predict subsequent development of delayed cardiotoxicity, are rarely of clinical importance, and are generally not a consideration for discontinuation of daunorubicin treatment.
Late (i.e. delayed) events.
Delayed cardiotoxicity usually develops late in the course of therapy with daunorubicin or within 2 to 3 months after treatment termination, but later events (several months to years after completion of treatment) have also been reported.
Delayed cardiomyopathy is manifested by reduced left ventricular ejection fraction (LVEF) and/or signs and symptoms of congestive heart failure (CHF) such as dyspnoea, pulmonary oedema, dependent oedema, cardiomegaly and hepatomegaly, oliguria, ascites, pleural effusion, and gallop rhythm. Life-threatening CHF is the most severe form of anthracycline-induced cardiomyopathy and represents the cumulative dose-limiting toxicity of the drug.
Risk factors for cardiotoxicity. There is increased risk of cardiac toxicity (and lower cumulative dosage limit) in patients previously treated with doxorubicin or in those who received prior or concomitant radiation therapy that encompassed the heart (mediastinal/pericardial area).
Pre-existing active or dormant heart disease, concomitant use of drugs with the ability to suppress cardiac contractility and previous therapy with other anthracyclines or anthracenediones are suspected co-factors of increased risk of daunorubicin-induced cardiac toxicity. It is probable that the toxicity of daunorubicin and other anthracyclines or anthracenediones is additive.
Anthracyclines including daunorubicin should not be administered in combination with other cardiotoxic agents (e.g. trastuzumab) unless the patient's cardiac function is closely monitored. Patients receiving anthracyclines after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab (variable half-life; washout period up to 7 months), may also be at an increased risk of developing cardiotoxicity. Under these conditions, a total cumulative dose of 400 mg/m2 in adults should be exceeded only with extreme caution.
It has also been suggested, but is not clearly established, that concurrent therapy with cyclophosphamide or some other antineoplastic agents (e.g. dacarbazine, dactinomycin, mitomycin) may increase the risk of daunorubicin-induced cardiotoxicity.
Cardiac function must be carefully monitored in all patients receiving high cumulative doses and in those with risk factors. However, cardiotoxicity with daunorubicin may occur at lower cumulative doses whether or not cardiac risk factors are present.
Total cumulative dosage. The incidence of cardiotoxicity is more frequent in adults receiving a total cumulative dose over 550 mg/m2 (or over 20 mg/kg body weight) or over 400 mg/m2 in patients who have received concurrent cyclophosphamide or previous chest irradiation, in the elderly, and in patients with a history of cardiac disease or mediastinal radiation. In adults, at total cumulative doses less than 550 mg/m2, acute congestive cardiac failure is seldom encountered, although rare instances of pericarditis/myocarditis, not dose related, have been reported. Also see Paediatric use later in this section.
Monitoring of cardiac function. Cardiac function should be assessed before patients undergo treatment with daunorubicin and must be monitored throughout therapy to minimise the risk of incurring severe cardiac impairment. There is no absolutely reliable method of predicting the patients in whom acute congestive heart failure will develop as a result of daunorubicin therapy. However, certain changes in the ECG and a decrease in the left ventricular ejection fraction (LVEF) from pretreatment baseline may help to recognise those patients at greatest risk. On the basis of the ECG, a decrease equal to or greater than 30% in limb lead QRS voltage has been associated with a significant risk of drug-induced cardiomyopathy. The appropriate quantitative method for repeated evaluation of LVEF includes multi-gated radionuclide angiography (MUGA) or echocardiography (ECHO).
A baseline cardiac evaluation with an ECG and either a MUGA scan or an ECHO is recommended, especially in patients with risk factors for increased cardiotoxicity. Repeated MUGA or ECHO determinations of LVEF should be performed, particularly with higher, cumulative anthracycline doses. The technique used for assessment should be consistent throughout follow-up.
The risk of cardiotoxicity may be decreased through regular monitoring of LVEF during the course of treatment with prompt discontinuation of daunorubicin at the first sign of impaired cardiac function. Early clinical diagnosis of drug-induced congestive heart failure appears to be essential for successful treatment with digoxin, diuretics, sodium restriction and bed rest.
Bone marrow depression.
Bone marrow depression and consequent marked cytopenia will occur in all patients who receive daunorubicin and requires careful monitoring. The severity being dependent on the dose received and the regenerative capacity of the bone marrow. Evaluation of response based on bone marrow status cellularity is necessary to guide daunorubicin treatment.
Myelosuppression is manifested primarily by leukopenia, which is usually severe, and thrombocytopenia. Anaemia may also occur. Leukocyte and platelet nadirs usually occur around days 10-14, with recovery around day 21 following therapy.
Haematologic profiles must be carefully assessed before and during each cycle of daunorubicin therapy, including differential white blood cell counts.
Clinical consequences of severe myelosuppression include fever, infection, sepsis/septicaemia, septic shock, haemorrhage, tissue hypoxia, or death. During a course of therapy special attention should be devoted to patients with severe neutropenia and fever (febrile neutropenia), a condition that can be possibly followed by septicaemia and death.
In a variable proportion of cases, a severe aplasia will develop which must be anticipated in every case by eliminating infection before treatment, by isolating the patient from infection during the treatment and by the use of supportive therapy, including the continuous administration of anti-infective agents, the administration of platelet rich plasma or fresh whole blood transfusion and, under some circumstances, the transfusion of blood or white cells from cases of hyperleucocytic chronic myeloid leukaemia. Therapy with daunorubicin should not be started in patients with pre-existing drug-induced bone marrow depression unless the benefit from such treatment warrants the risk.
Secondary leukaemia.
Secondary leukaemia, with or without a preleukaemic phase, has been reported in patients treated with anthracyclines, including daunorubicin. Secondary leukaemia is more common when such drugs are given in combination with DNA-damaging antineoplastic agents, in combination with radiotherapy, when patients have been heavily pre-treated with cytotoxic drugs, or when doses of the anthracyclines have been escalated. These leukaemias can have a 1 to 3-year latency period.
Immunosuppression/increased susceptibility to infections.
Daunorubicin possesses immunosuppressive properties. Appropriate measures should be taken to prevent secondary infection.
Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including daunorubicin, may result in serious or fatal infections. Vaccination with a live vaccine should be avoided in patients receiving daunorubicin. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions, Live virus vaccines).
Enhanced toxicity.
Daunorubicin may enhance the toxicity of other cytotoxic agents when administered concurrently and dosage should be suitably reduced (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Gastrointestinal effects.
Nausea and vomiting are usually mild and transient, occurring soon after administration and lasting 24 to 48 hours. Severe nausea and vomiting may produce dehydration. Nausea and vomiting may be prevented or alleviated by the administration of appropriate antiemetic therapy.
Mucositis (mainly stomatitis, less often oesophagitis) may occur in patients undergoing daunorubicin therapy. Mucositis/stomatitis generally appear early after drug administration (burning and erythema of the oral mucosa; sores in the mouth and/or lips occurring 3 to 7 days after administration) and if severe, may progress over a few days to mucosal ulcerations (7 to 10 days after administration). Most patients recover from this adverse event by the third week of therapy.
Effects at site of injection.
Phlebosclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Following the recommended administration procedures may minimise the risk of phlebitis/thrombophlebitis at the injection site (see Section 4.2 Dose and Method of Administration).
Extravasation.
Extravasation of daunorubicin at the site of intravenous administration can cause local pain, severe tissue lesions (vesication, severe cellulitis) and necrosis. Should signs or symptoms of extravasation occur during intravenous administration of daunorubicin, the drug infusion should be immediately stopped.
Hyperuricaemia/tumour lysis syndrome.
Daunorubicin may induce hyperuricaemia as a consequence of the extensive purine catabolism that accompanies rapid drug-induced destruction of a large number of leukaemia cells (tumour lysis syndrome).
It is recommended to check the blood uric acid, urea, potassium, calcium phosphate and creatinine levels, three or four times a week during the first week of treatment. Hydration, urine alkalinisation and prophylaxis with allopurinol to prevent hyperuricaemia may minimise potential complications of tumour lysis syndrome.
Neurotoxic effects.
There is little evidence of neurotoxic effects.
Alopecia.
Complete alopecia involving beard growth and the scalp, axillary and pubic hair occurs almost always with full doses of daunorubicin. This side-effect may cause distress to patients but is usually reversible, with regrowth of hair, which usually occurs within two to three months from the termination of therapy.
Radiotherapy.
Increased radiation toxicities such as skin reactions and mucositis may result from concurrent radiotherapy and daunorubicin therapy.
Embryo-fetal toxicity.
Daunorubicin can cause genotoxicity. An effective method of contraception is required for both male and female patients of childbearing potential during treatment and for at least 27 weeks after final dose of daunorubicin. Male patients with female partners of childbearing potential should use contraception during treatment and for at least 14 weeks following final dose of daunorubicin. Patients desiring to have children after completion of therapy should be advised to obtain genetic counselling if appropriate and available (see Section 4.6 Fertility, Pregnancy and Lactation; Section 5.3 Preclinical Safety Data).
Use in hepatic impairment.
Hepatotoxic effects have been reported resulting from daunorubicin treatment. Care should therefore be exercised when treating patients with impaired liver function.
The major route of elimination of daunorubicin is the hepatobiliary system. Serum total bilirubin should be evaluated before and during treatment with daunorubicin. Patients with elevated bilirubin may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients (see Section 4.2 Dose and Method of Administration, Dosage adjustment).
Patients with severe hepatic impairment must not receive daunorubicin (see Section 4.3 Contraindications).
Use in renal impairment.
Renal impairment can enhance the toxicity of recommended doses of daunorubicin. Prior to administration, it is recommended that renal function be evaluated using conventional clinical laboratory tests. Dosage should be reduced in patients with impaired renal function (see Section 4.3 Contraindications; Section 4.2 Dose and Method of Administration, Dosage adjustment).
Daunorubicin has been implicated as causing renal failure and should therefore be used with caution when renal damage exists.
Use in the elderly.
See Section 4.4 Special Warnings and Precautions for Use, Cardiac toxicity, Total cumulative dosage for information on incidence of cardiotoxicity in the elderly.
Paediatric use.
Infants and paediatrics appear to have a greater susceptibility to anthracycline-induced cardiac toxicity, and a long-term periodic evaluation of cardiac function has to be performed.
Paediatric patients may be more susceptible to the development of cardiomyopathy than adults. However, the risk of developing cardiomyopathy is reduced with a total dose less than 300 mg/m2 in paediatric patients over 2 years of age or at a total dosage of less than 10 mg/kg in paediatric patients younger than 2 years of age with a body surface area of less than 0.5 m2.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Daunorubicin is mainly used in combination with other cytotoxic drugs. Additive toxicity may occur especially with regard to bone marrow/haematologic and gastrointestinal effects (see Section 4.4 Special Warnings and Precautions for Use). The use of daunorubicin in combination chemotherapy with other potentially cardiotoxic drugs as well as the concomitant use of other cardioactive compounds (e.g. calcium channel blockers) requires monitoring of cardiac function throughout treatment. Changes in hepatic or renal function induced by concomitant therapies may affect daunorubicin metabolism, pharmacokinetics, therapeutic efficacy and/or toxicity.
Cyclophosphamide.
The cardiotoxic effects of daunorubicin may be enhanced by concurrent treatment with cyclophosphamide. Daunorubicin may exacerbate cyclophosphamide induced haemorrhagic cystitis. It is recommended that the total dose of daunorubicin not exceed 400 mg/m2 of body surface area when administered concurrently with cyclophosphamide.
Doxorubicin.
Previous treatment with doxorubicin increases the risk of daunorubicin induced cardiotoxicity. Daunorubicin should not be administered to patients who have received the complete cumulative dose of doxorubicin.
Allopurinol, colchicine, probenecid or sulphinpyrazone.
Daunorubicin may raise the concentration of uric acid in the blood. Control of hyperuricaemia and gout may require dosage adjustments to be made for antigout medications for better control. Allopurinol may be preferred to prevent or reverse daunorubicin induced hyperuricaemia because of the risk of uric acid nephropathy with uricosuric antigout agents.
Other bone marrow depressants.
Reduced dosage of daunorubicin may be required.
Hepatotoxic medications.
Concurrent administration may increase the risk of hepatotoxicity.
Live virus vaccines.
Due to its immunosuppressive properties, concurrent use of daunorubicin with a live virus vaccine may potentiate the replication of the vaccine, increase the adverse effects of the vaccine virus, or decrease the patient's antibody response to the virus.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Daunorubicin could induce chromosomal damage in human spermatozoa. Men undergoing treatment with daunorubicin should use effective contraceptive methods. Both men and women should seek advice on fertility preservation before treatment. In animal studies, daunorubicin caused testicular atrophy and total aplasia of spermatocytes in the seminiferous tubules in dogs.
(Category D)
Daunorubicin should not be used during pregnancy (see Section 4.3 Contraindications).
Daunorubicin has shown teratogenic, mutagenic and carcinogenic potential in animals. The drug must be considered as a potential cause of fetal malformations when administered to a pregnant woman.
Daunorubicin should only be used in women of childbearing potential if the expected benefits outweigh the risks of therapy and adequate contraception is used. If the patient becomes pregnant whilst receiving the drug, she should be advised of the potential hazard to the fetus.
Contraception in males and females.
Women of childbearing potential should use effective contraception during treatment with daunorubicin and for at least 27 weeks following the final dose. Male patients with female partners of childbearing potential should be advised to use effective contraception during treatment with daunorubicin and for at least 14 weeks after the final dose.
It is not known whether daunorubicin is excreted in breast milk. Because many drugs, including other anthracyclines, are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from daunorubicin, advise lactating women not to breastfeed during treatment with daunorubicin and for at least 6 days after last dose.4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
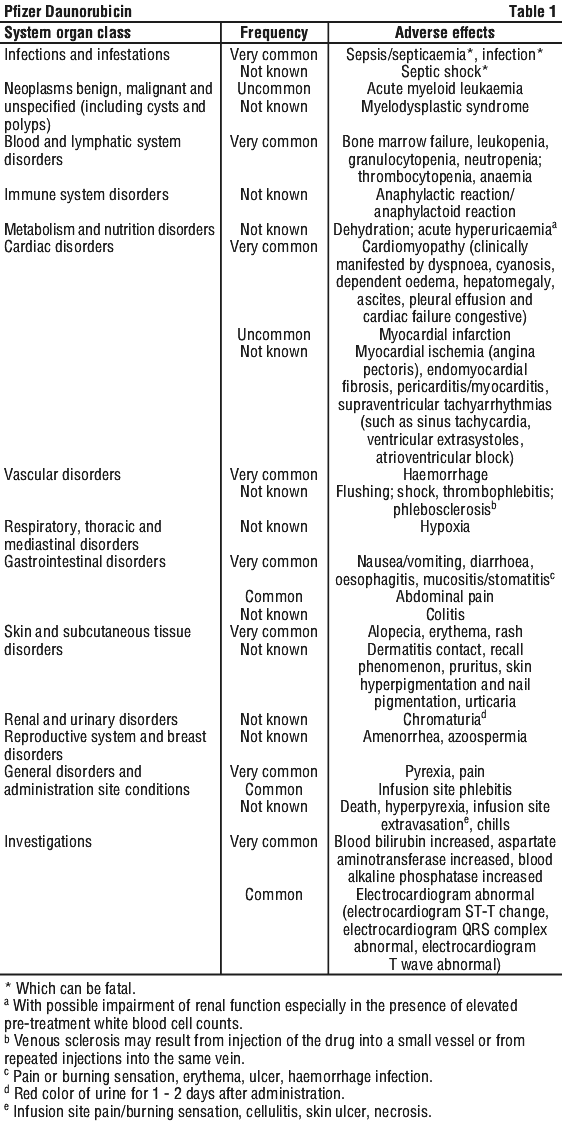
4.8 Adverse Effects (Undesirable Effects)
The adverse effects are listed by system organ class and frequency category. Frequency categories are defined as: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), not known (cannot be estimated from the available data) (also see Section 4.4 Special Warnings and Precautions for Use).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Signs and symptoms.
Clinical features of overdosage are likely to be an extension of daunorubicin's pharmacological action. Possible symptoms of toxicity are those listed under Section 4.8 Adverse Effects (Undesirable Effects).
Acute overdosage with daunorubicin will result in severe myelosuppression (mainly leukopenia and thrombocytopenia), gastrointestinal toxic effects (mainly mucositis) and acute cardiac complications.
Very high single doses of daunorubicin may be expected to cause acute myocardial degeneration (within 24 hours) and severe myelosuppression (within 10-14 days).
Management of overdosage.
Symptomatic supportive measures should be instituted. Particular attention should be given to prevention and treatment of possible severe haemorrhages or infections secondary to severe, persistent bone marrow depression.
Delayed cardiac failures have been observed with anthracyclines up to 6 months after an overdose. Patients have to be observed carefully and if signs of cardiac failure arise, they should be treated along conventional lines.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Class of drug.
Cytotoxic anthracycline antibiotic.
Mechanism of action.
Daunorubicin is an antineoplastic antibiotic which is structurally related to doxorubicin. The drug appears to act by inhibiting DNA and DNA-dependent RNA synthesis by forming a complex with DNA with intercalation between base pairs and uncoiling of the helix. Daunorubicin may also inhibit polymerase activity, affect regulation of gene expression, and be involved in free radical damage to DNA.
The drug is not cell cycle-phase specific although maximum cytotoxic activity occurs in the S phase. Daunorubicin also has antibacterial and immunosuppressive properties.
Clinical trials.
No data available.
5.2 Pharmacokinetic Properties
Distribution.
Daunorubicin is rapidly and widely distributed in tissues, with highest levels in the heart, kidneys, liver, lungs and spleen. It binds inside the cells to cellular components, mainly nucleic acids.
Daunorubicin does not cross the blood-brain barrier but appears to cross the placenta. It is not known if daunorubicin is present in breast milk.
Metabolism.
Daunorubicin is extensively metabolised in the liver and other tissues, mainly by cytoplasmic aldo-keto reductases, producing daunorubicinol, the major metabolite, which has antineoplastic activity. Approximately 40% of the drug in the plasma is present as daunorubicinol within 30 minutes and 60% in 4 hours after a dose of daunorubicin. Additional metabolism by reductive cleavage of the glycosidic bond produces aglycones, which have little or no cytotoxic activity and are demethylated and conjugated with sulphate and glucuronide by microsomal enzymes.
Daunorubicin metabolism may be altered in patients with impaired hepatic function.
Excretion.
Following rapid IV administration, total plasma concentrations of daunorubicin and its metabolites decline in a triphasic manner and plasma concentrations of unchanged daunorubicin decline in a biphasic manner.
The plasma half-life of daunorubicin averages 45 minutes in the initial phase and 18.5 hours in the terminal phase. By 1 hour after administration of daunorubicin, the predominant form of the drug in plasma is the metabolite daunorubicinol, which has as average terminal plasma half life of 26.7 hours.
Daunorubicin and its metabolites are excreted in the urine and bile, with urinary excretion accounting for 14-23% of the dose. Most urinary excretion of daunorubicin occurs within 3 days. After the first 24 hours, the drug is excreted in urine mainly as daunorubicinol. An estimated 40% of a dose is eliminated by biliary excretion.
5.3 Preclinical Safety Data
Genotoxicity.
Previous studies have demonstrated that daunorubicin was mutagenic (bacterial assay, V79 hamster cell assay). Daunorubicin was clastogenic in yeast and in human lymphocytes in vitro. In addition, dose dependent chromosomal aberrations were observed in the bone marrow of mice at doses ≥ 2.5 mg/kg after a single intravenous administration of daunorubicin (5/sex; 2.5, 5, 10 mg/kg IV).
Carcinogenicity.
Daunorubicin is potentially carcinogenic.6 Pharmaceutical Particulars
6.1 List of Excipients
Mannitol.
6.2 Incompatibilities
Heparin sodium and aluminium are incompatible with daunorubicin and will precipitate in solution. Incompatibility has also been reported when a daunorubicin hydrochloride solution is mixed with a solution of dexamethasone sodium phosphate, aztreonam, allopurinol sodium, fludarabine and piperacillin/tazobactam. Daunorubicin can be used in combination with other cytotoxic agents, but it is not recommended that it be mixed with other drugs in the same syringe.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Unopened vials.
Store below 25°C. Keep the container in the outer carton to protect from light.
Reconstituted solution.
To reduce microbiological hazard, use as soon as practicable after reconstitution/preparation. If storage is necessary, hold at 2-8°C for not more than 24 hours.
6.5 Nature and Contents of Container
Pfizer Daunorubicin Powder for Injection 18.7 mg daunorubicin (as hydrochloride) is supplied in glass vials in packs of 1 vial.
6.6 Special Precautions for Disposal
Only professionals, who have been trained in the safe use of the preparation of chemotherapeutic agents, should prepare daunorubicin for administration.
Operations such as transfer to syringes should only be carried out in the designated area. The personnel carrying out these procedures should be adequately protected with clothing, gloves and eye shields.
Pregnant personnel are advised not to handle cytotoxic agents.
Where the powder or reconstituted solution accidentally contacts skin or mucosa, the affected area should be immediately washed thoroughly with soap and water.
Luer-Lock fitting syringes are recommended.
All cleaning materials, all items used to dilute and administer daunorubicin or articles associated with body waste should be disposed of by placing in a thick polyethylene bag and incinerating at 1100°C.
Spills and disposal.
If spills occur, restrict access to the affected area. Wear two pairs of gloves (latex rubber), a respirator mask, a protective gown and safety glasses. Limit the spread of the spill by covering with absorbent material such as absorbent towel or adsorbent granules. Spills may also be treated with 3 M sulphuric acid and 0.3 M potassium permanganate (2:1) or 5% sodium hypochlorite.
Collect up the towel of absorbent/adsorbent material and other debris from spill and place in a leak proof plastic container labelled accordingly. Cytotoxic waste should be regarded as hazardous or toxic and clearly labelled 'Cytotoxic waste for incineration at 1100°C'. Waste material should be incinerated for at least one second at 1100°C. Cleanse the remaining spill area with copious amounts of water.6.7 Physicochemical Properties
Chemical structure.
 The chemical name is (8S,10S)-8-Acetyl-10-[(3-amino-2,3,6-trideoxy-α-l-lyxo-hexopyranosyl)oxy]-6,8,11-trihydroxy-1 methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione hydrochloride.
The chemical name is (8S,10S)-8-Acetyl-10-[(3-amino-2,3,6-trideoxy-α-l-lyxo-hexopyranosyl)oxy]-6,8,11-trihydroxy-1 methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione hydrochloride.
Molecular formula is C27H30ClNO10 and the molecular weight: 564.0.
Daunorubicin hydrochloride occurs as a hygroscopic, crystalline, orange-red powder, freely soluble in water and in methanol, slightly soluble in alcohol and practically insoluble in acetone.
CAS number.
23541-50-6.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.