1 Name of Medicine
Deferasirox.
2 Qualitative and Quantitative Composition
Deferasirox is a white to slightly yellow powder and is a non-chiral compound. At the physiological pH of the intestine, the solubility is about 40 mg/L. It is practically insoluble in water and across the pH range 1.2 - 6.00, and soluble at pH 8.00.
Pharmacor Deferasirox FC 90 mg.
Each film-coated tablet contains 90 mg deferasirox.
Pharmacor Deferasirox FC 180 mg.
Each film-coated tablet contains 180 mg deferasirox.
Pharmacor Deferasirox FC 360 mg.
Each film-coated tablet contains 360 mg deferasirox.
Excipient with known effect.
Not applicable.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Film-coated tablet.
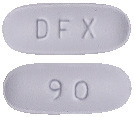
Pharmacor Deferasirox FC 90 mg.
Light blue color, film coated oval shaped biconvex tablets, debossed with 'DFX' on the one side and '90' on other side.
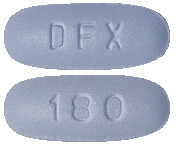
Pharmacor Deferasirox FC 180 mg.
Blue color, film coated oval shaped biconvex tablets, debossed with 'DFX' on the one side and '180' on other side.
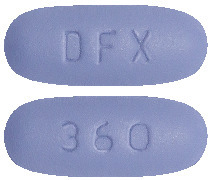
Pharmacor Deferasirox FC 360 mg.
Dark blue color, film coated, oval shaped, biconvex tablets, debossed with 'DFX' on the one side and '360' on other side.4.1 Therapeutic Indications
The treatment of chronic iron overload due to blood transfusions (transfusional haemosiderosis) in adults and paediatric patients 6 years and older. Pharmacor Deferasirox FC is also indicated for the treatment of chronic iron overload in paediatric patients aged 2 to 5 years who are unable to take desferrioxamine therapy or in whom desferrioxamine has proven ineffective.
Pharmacor Deferasirox FC is also indicated for the treatment of chronic iron overload in patients with non-transfusion-dependent thalassemia syndromes aged 10 years and older.
4.2 Dose and Method of Administration
Dosage.
Transfusional iron overload (Pharmacor Deferasirox FC film-coated tablets).
It is recommended that therapy with Pharmacor Deferasirox FC be started after the transfusion of approximately 20 units (about 100 mL/kg) of packed red blood cells or when there is evidence from clinical monitoring that chronic iron overload is present (e.g. serum ferritin > 1000 microgram/L). Doses (in mg/kg) must be calculated and rounded to the nearest whole tablet size. Pharmacor Deferasirox FC is available in three tablet strengths (90 mg, 180 mg and 360 mg). Dosing recommendations are the same for adult, paediatric and elderly patients.
The goals of iron chelation therapy are to remove the amount of iron administered in transfusions and, as required, to reduce the existing iron burden.
Pharmacor Deferasirox FC film-coated tablets are a strength-adjusted formulation of deferasirox with higher bioavailability compared to deferasirox dispersible tablet formulations (see Section 5.2 Pharmacokinetic Properties). For patients who are currently on chelation therapy with deferasirox dispersible tablet and switching to Pharmacor Deferasirox FC, the dose of Pharmacor Deferasirox FC film-coated tablets should be 30% lower than the dose of deferasirox dispersible tablet, rounded to the nearest whole tablet as shown in Table 3.
Starting dose.
The recommended initial daily dose of Pharmacor Deferasirox FC is 14 mg/kg body weight.
An initial daily dose of 21 mg/kg may be considered for patients receiving more than 14 mL/kg/month of packed red blood cells (approximately > 4 units/month for an adult), and for whom the objective is reduction of iron overload.
An initial daily dose of 7 mg/kg may be considered for patients receiving less than 7 mL/kg/month of packed red blood cells (approximately < 2 units/month for an adult), and for whom the objective is maintenance of the body iron level.
For patients already well-managed on treatment with desferrioxamine, a starting dose of Pharmacor Deferasirox FC that is numerically one third of the desferrioxamine dose could be considered as shown in Tables 1 and 3 (e.g. a patient receiving 40 mg/kg/day of desferrioxamine for 5 days per week (or equivalent) could be transferred to a starting daily dose of 14 mg/kg/day of Pharmacor Deferasirox FC).
Dose adjustment.
It is recommended that serum ferritin be monitored every month and that the dose of Pharmacor Deferasirox FC is adjusted if necessary every 3 to 6 months based on the trends in serum ferritin. Dose adjustments may be made in steps of 3.5 to 7 mg/kg and are to be tailored to the individual patient's response and therapeutic goals (maintenance or reduction of iron burden). In patients not adequately controlled with doses of 21 mg/kg (e.g. serum ferritin levels persistently above 2500 microgram/L and not showing a decreasing trend over time), doses of up to 28 mg/kg may be considered. Doses above 28 mg/kg are not recommended because there is only limited experience with doses above this level.
In patients whose serum ferritin level has reached the target (usually between 500 and 1,000 microgram/L), dose reductions in steps of 3.5 to 7 mg/kg should be considered to maintain serum ferritin levels within the target range and to minimise the risk of overchelation (see Section 4.4 Special Warnings and Precautions for Use). If serum ferritin falls consistently below 500 microgram/L, an interruption of treatment should be considered. As with other iron chelator treatment, the risk of toxicity of Pharmacor Deferasirox FC may be increased when inappropriately high doses are given in patients with a low iron burden or with serum ferritin levels that are only slightly elevated (see Section 4.4 Special Warnings and Precautions for Use).
In children aged between 2 and 5 years, exposure to deferasirox is lower than in adults. This age group may therefore require higher maintenance doses than adults. However, the initial dose should be the same as in adults, followed by individual titration.
The corresponding recommended doses for both formulations are shown in Table 1.

Non-transfusion-dependent thalassemia (NTDT) syndromes (Pharmacor Deferasirox FC film-coated tablets).
Dosage.
Chelation therapy should only be initiated when there is evidence of iron overload (liver iron concentration (LIC) ≥ 5 mg Fe/g dry weight (dw) or serum ferritin consistently > 800 microgram/L). In patients with no LIC assessment, caution should be taken during chelation therapy to minimize the risk of over-chelation.
Pharmacor Deferasirox FC film-coated tablets are a strength-adjusted formulation of deferasirox with higher bioavailability compared to deferasirox dispersible tablet formulation (see Section 5.2 Pharmacokinetic Properties). For patients who are currently on chelation therapy with deferasirox dispersible tablet and switching to Pharmacor Deferasirox FC, the dose of Pharmacor Deferasirox FC film-coated tablets should be 30% lower than the dose of deferasirox dispersible tablet, rounded to the nearest whole tablet.
Starting dose.
The recommended initial daily dose of Pharmacor Deferasirox FC is 7 mg/kg body weight.
Dose adjustment.
It is recommended that serum ferritin be monitored every month to assess the patient's response to therapy and to minimise the risk of overchelation (see Section 4.4 Special Warnings and Precautions for Use). Every 3 to 6 months of treatment, consider a dose increase in increments of 3.5 to 7 mg/kg if the patient's LIC is ≥ 7 mg Fe/g dw, or serum ferritin is consistently > 2,000 microgram/L and not showing a downward trend, and the patient is tolerating the drug well. Doses above 14 mg/kg are not recommended because there is no experience with doses above this level in patients with non-transfusion-dependent thalassemia syndromes.
In patients in whom LIC was not assessed and serum ferritin is ≤ 2,000 microgram/L, dosing should not exceed 7 mg/kg.
For patients in whom the dose was increased to > 7 mg/kg, dose reduction is recommended to 7 mg/kg or less when LIC is < 7 mg Fe/g dw or serum ferritin is ≤ 2,000 microgram/L.
Once a satisfactory body iron level has been achieved (LIC < 3 mg Fe/g dw or serum ferritin < 300 microgram/L), treatment should be interrupted. Treatment should be re-initiated when there is evidence from clinical monitoring that chronic iron overload is present.
The corresponding recommended doses for both formulations are shown in Table 2.

Transfusional iron overload and non-transfusion-dependent thalassemia syndromes.
Information on dose conversion between dispersible tablets and film-coated tablets, as well as desferrioxamine is shown in Table 3.

Method of administration.
The film-coated tablets should be swallowed whole with some water. For patients who are unable to swallow whole tablets, Pharmacor Deferasirox FC film-coated tablets may be crushed and administered by sprinkling the full dose on soft food like yogurt or apple sauce (apple puree). The dose should be immediately and completely consumed, and not stored for future use. Pharmacor Deferasirox FC should be taken once a day, preferably at the same time each day, and may be taken on an empty stomach or with a light meal.
The film-coated tablets should not be taken with a high fat meal (see Section 5.2 Pharmacokinetic Properties).
Special populations.
Patients with renal impairment.
Patients with pre-existing renal conditions, or patients who are receiving medicinal products that may depress renal function may be more at risk of complications. Therefore serum creatinine and/or creatinine clearance should be monitored weekly in the first month after initiation or modification of therapy (including switching formulation), and monthly thereafter. Caution should be used in patients with creatinine clearance between 40 and less than 90 mL/min, particularly in cases where there are additional risk factors that may impair renal function such as concomitant medications, dehydration, or severe infections. Reduce dose by 50% in patients with renal impairment (CrCl 40 - 60 mL/min) (see Section 4.3 Contraindications).
Consider dose reductions, interruptions, or discontinuation for increases in serum creatinine. For adult patients, the daily dose of Pharmacor Deferasirox FC may be reduced by 7 mg/kg if a non-progressive rise in serum creatinine by > 33% above the average of the pre-treatment measurements is seen at two consecutive visits, and cannot be attributed to other causes. For paediatric patients, the dose may be reduced by 7 mg/kg if serum creatinine levels rise above the age-appropriate upper limit of normal at two consecutive visits.
If there is a progressive increase in serum creatinine beyond the upper limit of normal, Pharmacor Deferasirox FC should be interrupted. Therapy with Pharmacor Deferasirox FC may be reinitiated depending on the individual clinical circumstances.
Patients with hepatic impairment.
For patients with moderate hepatic impairment (Child-Pugh B), the starting dose should be reduced by approximately 50%. Pharmacor Deferasirox FC should not be used in patients with severe hepatic impairment (Child-Pugh C) (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Paediatric patients.
The dosing recommendations for paediatric patients are the same as for adult patients. It is recommended that serum ferritin be monitored every month to assess the patient's response to therapy and to minimise the risk of overchelation (see Section 4.4 Special Warnings and Precautions for Use). Changes in weight of paediatric patients over time must be taken into account when calculating the dose (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Elderly patients.
The dosing recommendations for elderly patients are the same as described above. In clinical trials, elderly patients experienced a higher frequency of adverse reactions than younger patients and should be monitored closely for adverse reactions that may require a dose adjustment (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Patient monitoring.
Dosage adjustments in the presence of an adverse reaction.
Dosage adjustments are recommended for patients experiencing non-progressive elevations in serum creatinine, elevations in liver transaminases or those patients experiencing rash (see Section 4.4 Special Warnings and Precautions for Use).4.3 Contraindications
Creatinine clearance < 40 mL/min or serum creatinine > 2 times the age-appropriate upper limit of normal.
Platelet counts < 50 x 109/L.
High risk myelodysplastic syndrome (MDS) patients and patients with other haematological and non-haematological malignancies who are not expected to benefit from chelation therapy due to the rapid progression of their disease.
Hypersensitivity to the active substance or to any of the excipients listed, see Section 6.1.
4.4 Special Warnings and Precautions for Use
The decision to remove accumulated iron should be individualized based on anticipated clinical benefit and risks of chelation therapy (see Section 4.2 Dose and Method of Administration).
Caution should be used in elderly patients due to a higher frequency of adverse reactions. In clinical trials, elderly patients experienced a higher frequency of adverse reactions than younger patients and should be monitored closely for adverse reactions that may require a dose adjustment.
In post-marketing experience, there have been reports of serious adverse reactions, some with fatal outcome, in patients taking deferasirox therapy, predominantly when the drug was administered to patients with advanced age, complications from underlying conditions, or very advanced disease. Most of these deaths occurred within six months of deferasirox initiation, and generally involved worsening of the underlying condition. The reports do not rule out the possibility that deferasirox may have contributed to deaths.
Use in hepatic impairment.
Deferasirox has been studied in a clinical trial in subjects with hepatic impairment. For patients with moderate hepatic impairment (Child-Pugh B), the starting dose should be reduced by approximately 50%. Pharmacor Deferasirox FC should not be used in patients with severe hepatic impairment (Child-Pugh C) (see Section 5.2 Pharmacokinetic Properties). Deferasirox treatment has been initiated only in patients with baseline liver transaminase levels up to 5 times the upper limit of the normal range. The pharmacokinetics of deferasirox were not influenced by such transaminase levels. Deferasirox is principally eliminated by glucuronidation and is minimally (about 8%) metabolised by oxidative cytochrome P450 enzymes (see Section 5.2 Pharmacokinetic Properties).
Although uncommon (0.3%), elevations of transaminases greater than 10 times the upper limit of the normal range, suggestive of hepatitis, have been observed in clinical trials. There have been post-marketing reports of hepatic failure in patients treated with deferasirox. Most reports of hepatic failure occurred in patients greater than 55 years of age and in patients with significant comorbidities including liver cirrhosis and multi-organ failure. Fatal outcomes were reported in some of these patients (see Section 4.8 Adverse Effects (Undesirable Effects)). It is recommended that serum transaminases, bilirubin and alkaline phosphatase be monitored before the initiation of treatment, every 2 weeks during the first month and monthly thereafter. If there is a persistent and progressive increase in serum transaminase levels that cannot be attributed to other causes, Pharmacor Deferasirox FC should be interrupted. Once the cause of the liver function test abnormalities has been clarified or after return to normal levels, cautious re-initiation of Pharmacor Deferasirox FC treatment at a lower dose followed by gradual dose escalation may be considered.
Use in renal impairment.
Renal toxicity, renal failure, proteinuria.
Acute renal failure, fatal in some patients and requiring dialysis in others, has been reported following the post-marketing use of deferasirox (see Section 4.8 Adverse Effects (Undesirable Effects)). Most fatalities occurred in patients with multiple co-morbidities and who were in advanced stages of their haematological disorders. There is limited data on the molecular mechanism of nephrotoxicity of deferasirox.
It is recommended that serum creatinine and/or creatinine clearance be assessed in duplicate before initiating therapy, to establish a reliable pre-treatment baseline and monitored monthly thereafter.
Deferasirox has not been studied in patients with renal impairment and must be used with caution in such patients. Patients with pre-existing renal conditions, or patients who are receiving medicinal products that may depress renal function may be more at risk of complications. Therefore, serum creatinine and/or creatinine clearance should be monitored weekly in the first month after initiation or modification of therapy (including switching formulation), and monthly thereafter. Caution should be used in patients with creatinine clearance between 40 and less than 90 mL/min, particularly in cases where there are additional risk factors that may impair renal function such as concomitant medications, dehydration, or severe infections. Reduce dose by 50% in patients with renal impairment (CrCl 40 - 60 mL/min) (see Section 4.2 Dose and Method of Administration; Section 4.3 Contraindications).
Renal tubulopathy has been reported in patients treated with deferasirox. The majority of these patients were children and adolescents with beta-thalassaemia and serum ferritin levels < 1,500 microgram/L.
Dose reduction or interruption may be considered if abnormalities occur in levels of markers of renal tubular function and/or as clinically indicated.
Tests for proteinuria should be performed monthly.
Care should be taken to maintain adequate hydration in patients who develop diarrhoea or vomiting.
Largely non-progressive rises in serum creatinine have been noted in some patients treated with deferasirox, usually within the normal range. This has been observed in both paediatric and adult patients with iron overload during the first year of treatment.
A retrospective study was conducted in a subset of patients involved in deferasirox registration studies aiming to assess long term renal safety up to 13 years later. A total of 292 patients (80% of the original population) were studied (282 in Safety Set); more than 90% of patients had observations for at least 7 years.
Less than 2% of patients have been taking deferasirox exclusively from the time of the registration studies. During the retrospective period, the majority of patients (63.5% Safety Set) took at least 1 other iron-chelating therapy; deferoxamine (56.0% Safety Set) and deferiprone (48.2%). The other chelators were given in most cases sequentially.
Serum creatinine (the primary endpoint) was found to be stable through the retrospective study. The available data on other renal markers was sparse. Renal adverse events were reported in 30% of patients in the overall safety Set.
Consider dose reductions, interruptions, or discontinuation for increases in serum creatinine. For adult patients, the daily dose of Pharmacor Deferasirox FC may be reduced by 7 mg/kg if a non-progressive rise in serum creatinine by > 33% above the average of the pre-treatment measurements is seen at two consecutive visits, and cannot be attributed to other causes. For paediatric patients, the dose may be reduced by 7 mg/kg if serum creatinine levels rise above the age-appropriate upper limit of normal at two consecutive visits.
If there is a progressive increase in serum creatinine beyond the upper limit of normal, Pharmacor Deferasirox FC should be interrupted. Therapy with Pharmacor Deferasirox FC may be reinitiated depending on the individual clinical circumstances.
The recommendations for renal function monitoring are summarized in the Table 4.

Gastrointestinal.
Gastrointestinal irritation may occur during Pharmacor Deferasirox FC treatment. Upper gastrointestinal ulceration and haemorrhage have been reported in patients, including children and adolescents, receiving deferasirox. There have been rare reports of fatal GI haemorrhages, especially in elderly patients who had advanced hematologic malignancies and/or low platelet counts. Multiple ulcers have been observed in some patients (see Section 4.8 Adverse Effects (Undesirable Effects)). Physicians and patients should remain alert for signs and symptoms of GI ulceration and haemorrhage during Pharmacor Deferasirox FC therapy and promptly initiate additional evaluation and treatment if a serious GI adverse event is suspected. There have been reports of ulcers complicated with gastrointestinal perforation (including fatal outcome).
Caution should be exercised in patients who are taking Pharmacor Deferasirox FC in combination with drugs that have known ulcerogenic potential, such as NSAIDs, corticosteroids, or oral bisphosphonates and in patients receiving anticoagulants (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Skin disorders.
Severe cutaneous adverse reactions (SCARs) including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS) which could be life-threatening or fatal have been reported. Patients should be advised of the signs and symptoms of severe skin reactions, and be closely monitored. If any SCAR is suspected Pharmacor Deferasirox FC should be discontinued immediately and should not be reintroduced.
Rare cases of erythema multiforme have been reported during deferasirox film coated tablet treatment.
Skin rashes may appear during Pharmacor Deferasirox FC treatment. For rashes of mild to moderate severity, Pharmacor Deferasirox FC may be continued without dose adjustment, since the rash often resolves spontaneously. For more severe rash, where interruption of treatment may be necessary, Pharmacor Deferasirox FC may be reintroduced after resolution of the rash, at a lower dose followed by gradual dose escalation.
Hypersensitivity reactions.
Rare cases of serious hypersensitivity reactions (such as anaphylaxis and angioedema) have been reported in patients receiving deferasirox, with the onset of the reaction occurring in the majority of cases within the first month of treatment (see Section 4.8 Adverse Effects (Undesirable Effects)). If reactions are severe, Pharmacor Deferasirox FC should be discontinued and appropriate medical intervention instituted. Pharmacor Deferasirox FC should not be reintroduced in patients who have experienced previous hypersensitivity reactions on deferasirox due to the risk of anaphylactic shock.
Disturbances of vision and hearing.
Auditory (decreased hearing) and ocular (lens opacities, cataracts, elevations in intraocular pressure, and retinal disorders) disturbances have been reported with deferasirox treatment (see Section 4.8 Adverse Effects (Undesirable Effects)). Auditory and ophthalmic testing (including slit lamp examination and dilated fundoscopy) is recommended before the start of Pharmacor Deferasirox FC treatment and at regular intervals thereafter (every 12 months). If disturbances are noted, dose reduction or interruption may be considered.
Cytopenias.
There have been postmarketing reports (both spontaneous and from clinical trials) of cytopenias including agranulocytosis, neutropenia, thrombocytopenia, and pancytopenia in patients treated with deferasirox. Some of these patients died however the relationship of these episodes to treatment with deferasirox is uncertain. Most of these patients had preexisting haematologic disorders that are frequently associated with bone marrow failure (see Section 4.8 Adverse Effects (Undesirable Effects)). In line with the standard clinical management of such haematological disorders, blood counts should be monitored regularly.
Dose interruption of treatment with Pharmacor Deferasirox FC should be considered in patients who develop unexplained cytopenia. Reintroduction of therapy with Pharmacor Deferasirox FC may be considered, once the cause of the cytopenia has been elucidated.
Other precautions.
As with other iron chelator treatment, the risk of toxicity of Pharmacor Deferasirox FC may be increased when inappropriately high doses are given in patients with a low iron burden or with serum ferritin levels that are only slightly elevated. It is recommended that serum ferritin be measured every month in order to assess the patient's response to therapy and to avoid overchelation (see Section 4.2 Dose and Method of Administration). Closer monitoring of serum ferritin levels, as well as renal and hepatic function is recommended during periods of treatment with high doses and when serum ferritin levels are close to the target range. Dose reduction may be considered to avoid overchelation (see Section 4.2 Dose and Method of Administration). If serum ferritin falls consistently below 500 microgram/L, an interruption of treatment should be considered.
Pharmacor Deferasirox FC must not be combined with other iron chelator therapies as the safety of such combinations has not been established.
Use in the elderly.
In the elderly, data to support the safety and efficacy of doses greater than 30 mg/kg per day are limited. Closely monitor elderly patients for early signs or symptoms of adverse reactions that may require a dose adjustment. Elderly patients are at increased risk of Pharmacor Deferasirox FC toxicity due to the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Paediatric use.
Deferasirox has not been associated with growth retardation in children followed for up to 5 years in clinical trials with the dispersible tablet formulation. However, as a general precautionary measure, body weight and longitudinal growth in paediatric patients can be monitored at regular intervals (every 12 months). In children aged between 2 and 5 years, exposure to deferasirox is lower than in adults. This age group may therefore require higher maintenance doses than adults (see Section 4.2 Dose and Method of Administration).
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect on deferasirox on drug metabolizing enzymes.
Deferasirox inhibits human CYP3A4, CYP2C8, CYP1A2, CYP2A6, CYP2D6 and CYP2C19 in vitro. The clinical significance of deferasirox inhibition of CYP2A6, CYP2D6 and CYP2C19 is unknown.
Interaction with midazolam and other agents metabolised by CYP3A4.
In a healthy volunteer study, the concomitant administration of deferasirox dispersible tablet and midazolam (a CYP3A4 substrate) resulted in a decrease of midazolam exposure by 17%. In the clinical setting, this effect may be more pronounced. Therefore, due to a possible decrease in efficacy, caution should be exercised when deferasirox is combined with substances metabolised through CYP3A4 (e.g. ciclosporin, simvastatin, hormonal contraceptive agents).
Agents that may decrease Pharmacor Deferasirox FC systemic exposure.
In a healthy volunteer study, the concomitant administration of deferasirox (single dose of 30 mg/kg, dispersible tablet formulation) and the potent UDP-glucuronosyltransferase (UGT) inducer rifampicin (repeated dose of 600 mg/day) resulted in a decrease of deferasirox exposure by 44% (90% CI: 37% 51%). Therefore, the concomitant use of Pharmacor Deferasirox FC with potent UGT inducers (e.g. rifampicin, phenytoin, phenobarbital, ritonavir) may result in a decrease in Pharmacor Deferasirox FC efficacy. If Pharmacor Deferasirox FC and a potent UGT inducer are used concomitantly, increases in the dose of Pharmacor Deferasirox FC should be considered based on clinical response to therapy.
Interaction with repaglinide and other agents metabolised by CYP2C8.
In a healthy volunteer study, the concomitant administration of deferasirox (repeated dose of 30 mg/kg/day, dispersible tablet formulation) and the CYP2C8 substrate repaglinide (single dose of 0.5 mg) resulted in an increase in repaglinide AUC and Cmax by 131% (90% CI: 103% 164%) and 62% (90% CI: 42% 84%), respectively. When deferasirox and repaglinide are used concomitantly, careful monitoring of glucose levels should be performed. An interaction between deferasirox and other CYP2C8 substrates like paclitaxel cannot be excluded.
Interaction with theophylline and other agents metabolized by CYP1A2.
In a healthy volunteer study, the concomitant administration of deferasirox (repeated dose of 30 mg/kg/day, dispersible tablet formulation) and the CYP1A2 substrate theophylline (single dose of 120 mg) resulted in an increase in theophylline AUC by 84% (90% CI: 73% to 95%). The single dose Cmax was not affected, but an increase of theophylline Cmax is expected to occur with chronic dosing. Therefore, the concomitant use of deferasirox with theophylline is not recommended. When deferasirox and theophylline are used concomitantly, monitoring of theophylline concentration and possible theophylline dose reduction should be considered.
For substances that are predominantly metabolised by CYP1A2 and that have narrow therapeutic index (e.g. clozapine, tizanidine), the same recommendations apply as for theophylline. An interaction between deferasirox and other CYP1A2 substrates cannot be excluded. Use caution when Pharmacor Deferasirox FC is administered with other drugs metabolized by CYP1A2 such as clozapine, cyclobenzaprine, imipramine, haloperidol, fluvoxamine, mexiletine, naproxen, olanzapine, riluzole, tacrine, tizanidine, zileuton and zolmitriptan.
Interaction with busulfan.
Based on literature reports, concomitant administration of deferasirox and busulfan resulted in an increase of busulfan exposure (AUC). The mechanism of the interaction remains unclear. Caution should be exercised when deferasirox is combined with busulfan and the patient's plasma concentrations of busulfan should be monitored.
Other information.
No interaction was observed between deferasirox and digoxin in healthy volunteers. The concomitant administration of deferasirox and vitamin C has not been formally studied. Doses of vitamin C up to 200 mg per day have not been associated with adverse consequences.
The concomitant use of deferasirox with cholestyramine may result in a decrease in Pharmacor Deferasirox FC efficacy. In a study in healthy volunteers, the administration of cholestyramine after a single dose of deferasirox resulted in a 45% decrease in deferasirox AUC.
Anticipated interactions resulting in a concomitant use not recommended.
The concomitant administration of deferasirox and aluminium-containing antacid preparations has not been formally studied. Although deferasirox has a lower affinity for aluminium than for iron, deferasirox must not be taken with aluminium-containing antacid preparations.
Concomitant administration of deferasirox with drugs that have known ulcerogenic potential, such as NSAIDs, corticosteroids, or oral bisphosphonates, and use of deferasirox in patients receiving anticoagulants may increase the risk of gastrointestinal irritation (see Section 4.4 Special Warnings and Precautions for Use).4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Fertility was unaffected in rats with doses of up to 75 mg/kg/day which resulted in a drug exposure (plasma AUC) that was less than the maximum human value.
(Category C)
Deferasirox was not teratogenic in rats or rabbits treated with doses up to and exceeding the maximum tolerated doses. Fetal developmental impairment and increased skeletal variations were seen in rats at a maternotoxic dose of 100 mg/kg/day which achieved a drug exposure (plasma AUC) that was similar to the maximum human value. No adverse effects on fetal development were observed in rabbits at a maternotoxic dose of 50 mg/kg/day which achieved a drug exposure about 30% of the maximum human value. Treatment of rats with a maternotoxic dose of 90 mg/kg/day from early gestation to end of lactation resulted in increased stillborn pups and reduced pup birthweight. No effect was seen with 30 mg/kg/day which achieved a drug exposure about 20% of the maximum human value.
Adequate and well-controlled studies in pregnant women have not been conducted. Deferasirox should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Caution should be exercised when deferasirox is combined with hormonal contraceptive agents that are metabolised through CYP3A4 due to a possible decrease in efficacy of contraceptive agents (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
It is not known if deferasirox is secreted into human milk. In an animal study, deferasirox was present in the milk of rats at higher concentration than in maternal plasma. Because many drugs are excreted in human milk, women should be advised against breast-feeding while taking deferasirox.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects of Pharmacor Deferasirox FC on the ability to drive or use machines have been performed. Patients experiencing the uncommon adverse effect of dizziness should exercise caution when driving or operating machinery (see Section 4.8 Adverse Effects (Undesirable Effects)).
4.8 Adverse Effects (Undesirable Effects)
In clinical trials in patients with transfusional iron overload, the most frequent reactions reported during chronic treatment with the deferasirox dispersible tablet formulation in adult and paediatric patients include gastrointestinal disturbances in about 26% of patients (mainly nausea, vomiting, diarrhoea, or abdominal pain), and skin rash in about 7% of patients. These reactions are dose-dependent, mostly mild to moderate, generally transient and mostly resolve even if treatment is continued. Mild, non-progressive increases in serum creatinine, mostly within the normal range, occur in about 36% of patients. These are dose-dependent, often resolve spontaneously and can sometimes be alleviated by reducing the dose (see Section 4.4 Special Warnings and Precautions for Use).
In clinical trials of the deferasirox dispersible tablet formulation in patients with transfusional iron overload, elevations of liver transaminases were reported in about 2% of patients. These were not clearly dose-related and many of these patients had elevated levels prior to receiving deferasirox. Elevations of transaminases greater than 10 times the upper limit of the normal range, suggestive of hepatitis, were uncommon (0.3%). There have been postmarketing reports of hepatic failure in patients treated with deferasirox. Most reports of hepatic failure involved patients with significant comorbidities including liver cirrhosis and multi-organ failure; fatal outcomes were reported in some of these patients.
In a 1-year, randomized, double-blind, placebo-controlled study of the deferasirox dispersible tablet formulation in patients with non-transfusion-dependent thalassemia syndromes and iron overload, diarrhoea (9.1%), rash (9.1%), and nausea (7.3%) were the most frequent study drug-related adverse events reported by patients receiving 10 mg/kg/day (dispersible tablet formulation). Abnormal serum creatinine and creatinine clearance values were reported in 5.5% and 1.8%, respectively, of patients receiving 10 mg/kg/day deferasirox. Elevations of liver transaminases greater than 2 times the baseline and 5 times the upper limit of normal were reported in 1.8% of patients treated with 10 mg/kg/day (dispersible tablet formulation).
As with other iron chelator treatment, high-frequency hearing loss and lenticular opacities (early cataracts) have been uncommonly observed in patients treated with deferasirox (see Section 4.4 Special Warnings and Precautions for Use).
Adverse events in clinical trials.
The data in Table 5 displays the adverse events, regardless of causality, occurring in > 5% of patients in either treatment group in the primary efficacy study 0107 in which 296 β-thalassaemia patients were treated with deferasirox dispersible tablet and 290 patients received desferrioxamine as an active comparator.
 The type and frequency of adverse events observed in patients with sickle cell disease and other rare anaemias were similar to those observed in patients with β-thalassaemia. The adverse event profile in patients < 16 years of age was similar to that seen in adults, regardless of disease state.
The type and frequency of adverse events observed in patients with sickle cell disease and other rare anaemias were similar to those observed in patients with β-thalassaemia. The adverse event profile in patients < 16 years of age was similar to that seen in adults, regardless of disease state.
In 49 adult β-thalassaemia patients treated for greater than 1 year and up to 3 years, the type and frequency of adverse events was similar to that seen in patients treated for up to 1 year.
Adverse reactions with suspected relationship to product.
The following adverse drug reactions, listed in Table 6, have been reported in clinical studies following treatment with deferasirox dispersible tablet. Adverse reactions are ranked below using the following convention: very common (≥ 1/10); common (≥ 1/100, < 1/10); uncommon (≥ 1/1,000, < 1/100); rare (≥ 1/10,000, < 1/1,000); very rare (< 1/10,000). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
 An open label, randomised safety study F2201 was conducted with deferasirox film-coated and dispersible tablets in 173 patients with transfusion-dependent thalassemia (see Clinical trials).
An open label, randomised safety study F2201 was conducted with deferasirox film-coated and dispersible tablets in 173 patients with transfusion-dependent thalassemia (see Clinical trials).
A comparable safety profile for film-coated and dispersible tablets was observed; although there was increased incidence of serious adverse events in the FCT arm (18.4%) vs. DT arm (15.1%), and 'Renal disorders' groupings in the FCT arm (34.5% vs. 26.7%, respectively). Considering occurrences of SAEs, overall fewer occurrences in the FCT arm were seen (23 occurrences for 16 patients in the FCT arm versus 30 occurrences for 13 patients in the DT arm). The higher proportion of renal adverse events in the FCT arm was due to an imbalance in occurrence of proteinuria related events (proteinuria, urine protein/creatinine ration increased, urine protein/creatinine ratio abnormal, urine albumin/creatinine ratio increased). A post-hoc evaluation of patients with renal events (renal adverse events and abnormal laboratory parameters) revealed that more patients receiving FCT were started on doses above the protocol-recommended range (n = 23; 26.4%) than patients receiving DT (n = 8; 9.3%).
Spontaneously reported adverse reactions, presented in Table 7, are reported voluntarily and it is not always possible to reliably establish frequency or a causal relationship to drug exposure.

Description of selected adverse drug reactions.
Cytopenias.
There have been postmarketing reports (both spontaneous and from clinical trials) of cytopenias including neutropenia and thrombocytopenia and aggravated anaemia in patients treated with deferasirox. Most of these patients had pre-existing haematologic disorders that are frequently associated with bone marrow failure (see Section 4.4 Special Warnings and Precautions for Use). The relationship of these episodes to treatment with deferasirox is uncertain.
Pancreatitis.
Cases of serious acute pancreatitis were observed with and without documented underlying biliary conditions.
Paediatric population.
Renal tubulopathy has been reported in patients treated with deferasirox. The majority of these patients were children and adolescents with beta-thalassaemia and serum ferritin levels < 1,500 microgram/L.
In a 5-year observational study in which 267 children aged 2 to < 6 years (at enrolment) with transfusional hemosiderosis received deferasirox (dispersible tablets) there were no clinically meaningful differences in the safety and tolerability profile of deferasirox in paediatric patients aged 2 to < 6 years compared to the overall adult and older paediatric population. The proportion of patients with SCr > ULN at two consecutive visits at least seven days apart was 4.4%. Eight (3.1%) patients had serum creatinine increases of ˃ 33% and above the ULN observed on two consecutive measurements at least seven days apart. Elevation of alanine aminotransferase (ALT) greater than 5 times the ULN was reported in 4.3% of children. The most frequently observed AEs with reported suspected relationship to study drug were increase in ALT (21.1%), increase in aspartate aminotransferase (AST, 11.9%), vomiting (5.4%), rash (5.0%), increase in blood creatinine (3.8%), abdominal pain (3.1%) and diarrhoea (1.9%). Growth and sexual development were within normal limits for patients with available assessment data (approximately 50% of patients at 5 years), however no definite conclusion can be drawn from this study on the influence of deferasirox. As a general precautionary measure in the management of paediatric patients with transfusional iron overload, body weight, height and sexual development should be monitored prior to therapy and at regular intervals (every 12 months).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Symptoms.
Cases of overdose (2 to 3 times the prescribed dose for several weeks) have been reported. In one case, this resulted in subclinical hepatitis which resolved without long-term consequences after a dose interruption. Single doses of 80 mg/kg of the deferasirox dispersible tablet formulation (corresponding to a dose of 56 mg/kg Pharmacor Deferasirox FC) in iron overloaded thalassaemic patients have been tolerated, with only mild nausea and diarrhoea noted. Single doses up to 40 mg/kg of the deferasirox dispersible tablet formulation (corresponding to a dose of 28 mg/kg Pharmacor Deferasirox FC) in normal subjects have been well tolerated.
Acute signs of overdose may include nausea, vomiting, headache, abdominal pain and diarrhoea. Hepatic and renal disorders have been reported, including cases of liver enzyme and creatinine increased with recovery after treatment discontinuation. An erroneously administered single dose of 90 mg/kg led to Fanconi syndrome which resolved after treatment.
Treatment.
There is no specific antidote for deferasirox. Standard procedures for management of overdose (e.g. induction of emesis or gastric lavage) may be indicated as well as symptomatic treatment, as medically appropriate.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Deferasirox is an orally active chelator that is highly selective for iron (III). It is a tridentate ligand that binds iron with high affinity in a 2:1 ratio. Deferasirox promotes excretion of iron, primarily in the faeces. Deferasirox has very low affinity for zinc and copper and does not cause constant low serum levels of these metals.
In an iron balance metabolic study in iron overloaded adult thalassaemic patients, deferasirox at daily doses of 10, 20 and 40 mg/kg (dispersible tablet formulation) induced the mean net excretion of 0.119, 0.329, and 0.445 mg Fe/kg body weight/day, respectively.
Desferasirox has been investigated in adult and paediatric patients (aged 2 years and older) with chronic iron overload due to blood transfusions. The underlying conditions requiring transfusion included beta-thalassaemia, sickle cell disease, and other congenital and acquired anaemias (myelodysplastic syndromes, Diamond-Blackfan syndrome, aplastic anaemia and other very rare anaemias) (see Clinical trials).
In patients with non-transfusion-dependent thalassemia syndromes and iron overload, treatment with deferasirox at a dose of 10 mg/kg/day (dispersible tablet formulation) for one year led to a reduction in mean liver iron concentration from baseline by 3.80 mg Fe/g dw, while an increase of 0.38 mg Fe/g dw was observed in patients treated with placebo. In addition, treatment with desferasirox at a dose of 10 mg/kg/day for one year led to a reduction in mean serum ferritin from baseline by 222.0 microgram/L, while an increase of 114.5 microgram/L was observed in patients treated with placebo.
Clinical trials.
Clinical efficacy studies with deferasirox DT formulation.
The primary efficacy study, Study 0107, was an open-label, randomised, Phase III, active comparator control study to compare deferasirox dispersible tablets and desferrioxamine (Desferal) in patients with β-thalassaemia and transfusional haemosiderosis. Patients ≥ 2 years of age were randomised in a 1:1 ratio to receive either oral deferasirox at starting doses of 5, 10, 20 or 30 mg/kg once daily or subcutaneous desferrioxamine at starting doses of 20 to 60 mg/kg for at least 5 days per week based on LIC (liver iron concentration) at baseline (2 to 3, > 3 to 7, > 7 to 14 and > 14 mg Fe/g dw). Patients randomised to desferrioxamine who had LIC values < 7 mg Fe/g dw were permitted to continue on their prior desferrioxamine dose, even though the dose may have been higher than specified in the protocol. Consequently, the ratio of deferasirox to desferrioxamine doses for the two lower LIC categories was disproportionately low (1:4) compared to the two upper LIC categories (1:2).
Treatment duration was 12 months. LIC, an accepted indicator of total body iron burden, was assessed at baseline and after 12 months of therapy by liver biopsy or non-invasively by biomagnetic susceptometry. Success rate, the primary efficacy endpoint, was defined as a reduction in LIC of ≥ 3 mg Fe/g dw for baseline values ≥ 10 mg Fe/g dw, reduction of LIC to below 7 mg Fe/g dw for baseline values ≥ 7 and < 10 mg Fe/g dw, or maintenance or reduction for baseline values < 7 mg Fe/g dw. Deferasirox was to be declared non-inferior to desferrioxamine if the lower limit of the 95% confidence interval (two-sided) of the difference in success rates was above 15%.
The primary efficacy population consisted of 553 patients (deferasirox n=276; desferrioxamine n=277) who had LIC evaluated at baseline and 12 months, or who discontinued prior to 12 months due to an AE. Fifty-one percent of the patients were < 16 years of age. The overall success rates were 52.9% for deferasirox and 66.4% for desferrioxamine with a difference of -13.5 in success rates and a 95% CI of [-21.6, -5.4]. Non-inferiority to desferrioxamine was not achieved because the lower limit of the CI was below -15%. However, non-inferiority was demonstrated in a group of patients with baseline LIC levels ≥ 7 mg Fe/g dw who were allocated to the higher dose groups (deferasirox doses of 20 or 30 mg/kg and desferrioxamine doses of ≥ 35 mg/kg). For this group of patients, the success rates with deferasirox and desferrioxamine were 58.6% and 58.9%, respectively, and the lower limit of the 95% CI (10.2%) was above the non-inferiority threshold of 15%. This additional analysis was a post-hoc amendment to the protocol prior to database lock.
In patients with LIC ≥ 7 mg Fe/g dw who were treated with deferasirox 20 to 30 mg/kg per day a statistically significant reduction in LIC from baseline was observed (-5.3 ± 8.0 mg Fe/g dw, p < 0.001, t-test). This reduction in LIC was not statistically significantly different from that observed in the desferrioxamine treated patients (4.3 ± 5.8 mg Fe/g dw, p = 0.367). Dose dependent effects in serum ferritin and in the ratio of iron excretion/iron intake from deferasirox doses of 5 to 30 mg/kg were also observed (Table 8).
 Desferrioxamine appeared more effective in the 2 to 5 years age group although the difference was not statistically significant. Success rates in this age group were 71.4% (95% CI: 54.7% - 88.2%) with desferrioxamine and 42.9% (95% CI: 24.5 - 61.2) with deferasirox. Reduced efficacy in this age group may have been the result of reduced systemic exposure. For a given mg/kg dose of deferasirox, systemic exposure in children aged 2-5 is approximately 50% lower than in adults (see Section 5.2 Pharmacokinetic Properties; Section 4.2 Dose and Method of Administration).
Desferrioxamine appeared more effective in the 2 to 5 years age group although the difference was not statistically significant. Success rates in this age group were 71.4% (95% CI: 54.7% - 88.2%) with desferrioxamine and 42.9% (95% CI: 24.5 - 61.2) with deferasirox. Reduced efficacy in this age group may have been the result of reduced systemic exposure. For a given mg/kg dose of deferasirox, systemic exposure in children aged 2-5 is approximately 50% lower than in adults (see Section 5.2 Pharmacokinetic Properties; Section 4.2 Dose and Method of Administration).
The results of the primary efficacy study are supported by the second major efficacy study, Study 0108, an open-label, non-comparative, phase II trial of efficacy and safety of deferasirox dispersible tablets given for 1 year to patients with chronic anaemias and transfusional haemosiderosis unable to be treated with desferrioxamine. Similar to Study 0107, patients received 5, 10, 20, or 30 mg/kg per day of deferasirox based on baseline LIC. The primary endpoint was to demonstrate a success rate significantly greater than 50% with deferasirox.
A total of 184 patients were treated in this study: 85 patients with β-thalassaemia and 99 patients with other congenital or acquired anaemias (myelodysplastic syndromes, n=47; Diamond-Blackfan syndrome, n=30; other, n=22). Nineteen percent of patients were < 16 years of age and 16% were ≥ 65. Thirty seven patients had not received prior chelation therapy. In the total population, the success rate (50.5%) was not statistically significantly higher than 50%. However, in patients with LIC ≥ 7 mg Fe/g dw for whom both baseline and end of study LIC was available and who received deferasirox 20 to 30 mg/kg per day, the success rate was 58.5% [p=0.022 (50.3, 66.6)] and there was a statistically significant reduction in the absolute LIC from baseline to end of study (-5.5 ± 7.4 mg Fe/g dw, p < 0.001, t-test). There was also a dose dependent effect on serum ferritin and the ratio of iron excretion to iron intake from doses of 5 to 30 mg/kg per day.
In both of these year-long clinical studies, monthly monitoring of serum ferritin was shown to reflect changes in liver iron concentration and thus can be used to monitor response to therapy.
Study 0109 was an open-label, randomised, Phase II, active comparator control study to compare deferasirox dispersible tablets and desferrioxamine in patients with sickle cell disease and transfusional hemosiderosis. As in Study 0107, patients received 5, 10, 20, or 30 mg/kg per day of deferasirox or subcutaneous desferrioxamine at doses of 20 to 60 mg/kg for 5 days per week based on baseline LIC. The primary objective of this study was safety and tolerability (see Section 4.8 Adverse Effects (Undesirable Effects)). A total of 132 patients were treated with deferasirox and 63 patients with desferrioxamine. At the 12 month analysis, dose-dependent increases in the ratio of iron excretion to iron intake from doses of 5 to 30 mg/kg per day of deferasirox were observed.
Study 2209 was a randomized, double-blind, placebo-controlled study to compare deferasirox dispersible tablets and placebo was conducted in patients with non-transfusion-dependent thalassemia syndromes and iron overload. Patients ≥ 10 years of age were enrolled in the study in a 2:1:2:1 randomization to receive either deferasirox dispersible tablets 5 mg/kg/day or 10 mg/kg/day, or matching placebo.
Transfusion independency of the patients was confirmed by the fact that blood transfusions 6 months prior to study start were not allowed and patients were excluded if a regular transfusion program was anticipated during the study. Iron overload was diagnosed by a serum ferritin > 300 microgram/L at screening (two consecutive values at least 14 days apart from each other) and LIC ≥ 5 mg Fe/g dw measured by R2 MRI at screening. All patients with non-transfusion-dependent thalassemia syndromes were allowed with the exception of patients with HbS-variants or those whose clinical condition allowed phlebotomy.
In total, 166 patients were randomized. Demographics were well balanced. The main underlying disease was beta-thalassemia intermedia in 95 (57.2%) patients and HbE beta-thalassemia in 49 (29.5%) patients. The primary efficacy endpoint of change in liver iron concentration (LIC) from baseline to Week 52 was statistically significant in favour of both deferasirox treatment groups compared with placebo (Table 9). Furthermore, a statistically significant dose effect of deferasirox was observed in favour of the 10 mg/kg/day dispersible tablet dose.
 The primary efficacy result was supported by additional analyses which showed a clear dose-response effect; this was reflected by a greater percentage of patients with an LIC decrease of ≥ 3 mg Fe/g dw in the 10 mg/kg/day deferasirox dispersible tablet group compared to the 5 mg/kg/day group (56.4% versus 32.7%, respectively). In addition, a reduction of ≥ 30% in LIC between baseline and Week 52 was reported in approximately twice as many patients in the 10 mg/kg/day deferasirox dispersible tablet group (49.15%) compared to the 5 mg/kg/day group (25.5%).
The primary efficacy result was supported by additional analyses which showed a clear dose-response effect; this was reflected by a greater percentage of patients with an LIC decrease of ≥ 3 mg Fe/g dw in the 10 mg/kg/day deferasirox dispersible tablet group compared to the 5 mg/kg/day group (56.4% versus 32.7%, respectively). In addition, a reduction of ≥ 30% in LIC between baseline and Week 52 was reported in approximately twice as many patients in the 10 mg/kg/day deferasirox dispersible tablet group (49.15%) compared to the 5 mg/kg/day group (25.5%).
In clinical trials, deferasirox has been shown to reduce liver iron concentration and serum ferritin levels. Clinical trials to demonstrate increased survival or to confirm clinical benefit have not been completed.
Clinical studies with deferasirox FCT and DT formulations.
Study F2201, an open label, randomised trial assessed the safety of deferasirox film-coated and dispersible tablets in 173 patients including 21 paediatric patients (aged < 18 years) with transfusion-dependent thalassemia or myelodysplastic syndrome, who were treated for 24 weeks. The study was not powered for results based upon the paediatric subset.
Patients' compliance, palatability of medications, GI tolerance and patients' satisfaction with treatment were some of the secondary endpoints of the study.
A comparable safety profile for film-coated and dispersible tablets was observed, although there was increased incidence of serious adverse events in the FCT arm (18.4%) vs. DT arm (15.1%) and 'Renal disorders' groupings in the FCT arm (34.5% vs. 26.7%, respectively). Considering occurrences of SAEs, overall fewer occurrences in the FCT arm were seen (23 occurrences for 16 patients in the FCT arm versus 30 occurrences for 13 patients in the DT arm). The higher proportion of renal adverse events in the FCT arm was due to an imbalance in occurrence of proteinuria related events (proteinuria, urine protein/creatinine ration increased, urine protein/creatinine ratio abnormal, urine albumin/creatinine ratio increased). A post-hoc evaluation of patients with renal events (renal adverse events and abnormal laboratory parameters) revealed that more patients receiving FCT were started on doses above the protocol-recommended range (n = 23; 26.4%) than patients receiving DT (n = 8; 9.3%).
An increased adherence to treatment, higher patient satisfaction and better palatability were reported in the film-coated tablet arm.
5.2 Pharmacokinetic Properties
Pharmacor Deferasirox FC film-coated tablets are a strength-adjusted formulation of deferasirox due to higher bioavailability compared to the deferasirox dispersible tablet formulation.
The film-coated tablet formulation (360 mg strength) was equivalent to deferasirox dispersible tablets (500 mg strength) in PK study F2102 with respect to the mean area under the plasma concentration time curve (AUC) under fasting conditions. The Cmax was increased by 30% (90% CI: 20.3% - 40.0%); therefore FCT and DT formulations could not be claimed as being bioequivalent on prospectively defined bioequivalence criteria.
However, a post-hoc large clinical exposure-response analysis (study A2409) with the dispersible tablet formulation has revealed no evidence of clinically relevant effects of such Cmax increase.
This retrospective analysis was conducted at steady-state and used surrogate endpoints for AUC and Cmax (Ctrough and C2hours, respectively).
Absorption.
Deferasirox (dispersible tablet formulation) is rapidly absorbed following oral administration with a median time to maximum plasma concentration (Tmax) of about 1.5 to 4 hours. The absolute bioavailability (AUC) of deferasirox (dispersible tablet formulation) was about 70% (90% CI 0.62, 0.80) compared to an intravenous dose. The absolute bioavailability of the film-coated tablet formulation has not been determined. Bioavailability of deferasirox film-coated tablets was 36% greater than that with deferasirox dispersible tablets. The Cmax and AUC0-24h of deferasirox increase approximately linearly with dose under steady-state conditions. Upon daily oral dosing with the dispersible tablet formulation exposure increased by an accumulation factor of 1.3 to 2.3.
A food-effect study involving administration of the film-coated tablets to healthy volunteers under fasting conditions and with a low-fat (fat content < 10% of calories) or high-fat (fat content > 50% of calories) meal indicated that the AUC and Cmax were slightly decreased after a low-fat meal (by 11% and 16%, respectively). After a high-fat meal, AUC and Cmax were increased (by 18% and 29%, respectively). The increases in Cmax due to the change in formulation and due to the effect of a high-fat meal may be additive and therefore, it is recommended that Pharmacor Deferasirox FC should be taken either on an empty stomach or with a light meal (see Section 4.2 Dose and Method of Administration).
Distribution.
The in vitro binding of deferasirox to albumin (40 g/L) was constant at 98%-99% for deferasirox concentrations between 10 and 105 microgram/mL. Binding of deferasirox to α1-acid glycoprotein (1 g/L) decreased from 85% to 8% with increasing deferasirox concentrations (0.5-105 microgram/mL or 1.3 263 micromol/L), indicating saturable binding of this protein. Binding to γ-globulins was negligible. Overall, albumin is the main protein responsible for binding of deferasirox in plasma. Deferasirox has a small volume of distribution of approximately 14 L in adults.
Metabolism.
Glucuronidation is the main metabolic pathway for deferasirox, with subsequent biliary excretion. Deconjugation of glucuronidates in the intestine and subsequent reabsorption (enterohepatic recycling) is likely to occur. Deferasirox is mainly glucuronidated by UGT1A1 and to a lesser extent UGT1A3. CYP450-catalysed (oxidative) metabolism of deferasirox appears to be minor in humans (about 8%). No inhibition of deferasirox metabolism by hydroxyurea was observed in an in vitro study. Deferasirox undergoes enterohepatic recycling. In a healthy volunteer study, the administration of cholestyramine after a single dose of deferasirox resulted in a 45% decrease in deferasirox exposure (AUC).
Excretion.
Deferasirox and its metabolites are primarily excreted in the faeces (84% of the dose). Renal excretion of deferasirox and its metabolites is minimal (8% of the dose). The mean apparent elimination half-life (T1/2) after an oral dose ranged from 8 to 16 hours. Following IV administration deferasirox clearance was measured to be 3.53 ± 0.97 L/h.
Pharmacokinetics in special patient groups.
Children.
The overall exposure of adolescents (12 to ≤ 17 years) and children (2 to < 12 years) to deferasirox after single and multiple doses was lower than that in adult patients. In children younger than 6 years old exposure was about 50% lower than in adults (see Section 4.2 Dose and Method of Administration).
Elderly.
The pharmacokinetics of deferasirox has not been studied in elderly patients (aged 65 or older).
Gender.
Females have a moderately lower non-significant apparent clearance (by 17.5%) of deferasirox compared to males. Since dosing is individually adjusted according to response this is not expected to have clinical consequences.
Impaired renal or hepatic function.
The pharmacokinetics of deferasirox has not been studied in patients with renal impairment. The average AUC of deferasirox in 6 subjects with mild hepatic impairment (Child-Pugh A) increased 16% over that found in 6 subjects with normal hepatic function, while the average AUC of deferasirox in 6 subjects with moderate hepatic impairment (Child-Pugh B) increased 76% over that found in 6 subjects with normal hepatic function. The average Cmax of deferasirox in subjects with mild or moderate hepatic impairment increased 22% over that found in subjects with normal hepatic function. The impact of severe hepatic impairment (Child-Pugh C) was assessed in only one subject (see Section 4.4 Special Warnings and Precautions for Use). The pharmacokinetics of deferasirox was not influenced by liver transaminase levels up to 5 times the upper limit of the normal range.
5.3 Preclinical Safety Data
Genotoxicity.
Deferasirox was not genotoxic in in vitro tests for bacterial gene mutation or chromosomal aberrations in human lymphocytes. Positive responses were seen in an in vitro (V79) and in rat in vivo (bone marrow) micronucleus tests, which may have been related to iron chelation. No response was seen in another rat in vivo micronucleus test (liver) with doses that exceeded the maximum tolerated dose.
Carcinogenicity.
Deferasirox was not carcinogenic in a 104 week study in rats or in a 26 week study in transgenic p53 +/- heterozygous mice that were maintained on an iron-supplemented diet. The highest dose used in the rat study (60 mg/kg/day) resulted in a drug exposure (plasma AUC) that was about 15% of the maximum human value (at clinical dose of 30 mg/kg). In the mouse study, the highest doses of 200 mg/kg/day (males) and 300 mg/kg/day (females) resulted in drug exposures that were respectively slightly lower and slightly above the maximum human value.6 Pharmaceutical Particulars
6.1 List of Excipients
Each Pharmacor Deferasirox FC tablet contains:
Microcrystalline Cellulose, Colloidal anhydrous silica, Povidone, Crospovidone, Poloxamer, Hydrogenated Castor Oil, Sodium Hydroxide, Sodium Stearyl Fumarate, Opadry Complete Film Coating System 00F505006 Blue (for the 360 mg strength tablet), Opadry Complete Film Coating System 00F505007 Blue (for the 90 mg strength tablet), Opadry Complete Film Coating System 00F505008 Blue (for the 180 mg strength tablet).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C. Protect from moisture.
6.5 Nature and Contents of Container
Alu-Alu blister pack of 10's, 30's, 60's and 90's tablets.
HDPE bottle with polypropylene child-resistant closure of 30's and 90's tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.

CAS number.
201530-41-8.7 Medicine Schedule (Poisons Standard)
S4 - Prescription only medicine.
Summary Table of Changes