What is in this leaflet
This leaflet answers some common questions about Polivy. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Polivy against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Polivy is used for
Polivy contains the active ingredient called polatuzumab vedotin.
Polivy belongs to a group known as anti-neoplastic (or anti-cancer) agents.
Polivy is used to treat a type of cancer called diffuse large B-cell lymphoma.
It is used when the cancer has come back or has never responded to one or more previous treatments for this type of cancer and when you cannot receive a stem cell transplant
Polivy is made up of two substances:
- polatuzumab - a monoclonal antibody which recognises the large B-cell cancer cells
- vedotin - an anti-cancer substance
Polivy is designed to target and deliver the anti-cancer substance vedotin to the cancer cells to stop the growth and spread of the cancer cells.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
Before you are given Polivy
If you are not sure if you should start receiving Polivy, talk to your doctor
When you must not be given it
Do not take Polivy if you have an allergy to:
- polatuzumab vedotin or any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Before you start to take it
Tell your doctor if:
- you have ever had nerve problems such as numbness, tingling in the hands or feet or eyesight problems
- you have ever had liver problems such as hepatitis
- you think you may have an infection or have had long lasting or repeated infections
Tell your doctor right away if you become pregnant during treatment with Polivy.
Your doctor will advise you about using effective contraception to avoid becoming pregnant while you are being treated with Polivy and for 9 months after stopping treatment. If you are male, you should also use contraception with a female partner for 6 months after treatment with Polivy.
Discuss any future child bearing plans with your doctor before starting Polivy.
Tell your doctor if you are breastfeeding or plan to breastfeed. It is not known if Polivy passes into your breastmilk.
Do not breastfeed during treatment with Polivy for at least 3 months after the last dose/ treatment.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
If you have not told your doctor about any of the above, tell them before you are given Polivy.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
The following medicines may interfere with Polivy;
- Oral antifungal medications, e.g. ketoconazole, itraconazole, voriconazole
- Some antibiotics used to treat infections e.g. rifampicin
Your doctor and pharmacist have more information on medicines to be careful with or avoid while receiving Polivy.
How Polivy is given
Follow all directions given to you by your doctor or nurse carefully. They may differ from the information contained in this leaflet.
Polivy must be prepared by a healthcare professional and will be given in a hospital or clinic by a doctor or nurse.
Polivy is given by a drip into a vein (called an "intravenous infusion" or "IV").
The recommended dose is based on your weight and will be determined by your doctor.
Polivy is given by a slow drip into a vein (intravenous (IV) infusion) once every three weeks.
The first infusion will be given over 90 minutes. If the first infusion is well tolerated, your drip time may be shortened to 30 minutes.
The number of infusions you will be given depends on how you respond to treatment.
If you miss a dose
As Polivy is given under the supervision of your doctor, you are unlikely to miss a dose. However, if you forget or miss your appointment to receive Polivy, make another appointment as soon as possible. Do not wait for your next planned appointment. Your doctor will decide when your next dose of Polivy will be.
If you are given too much (overdose)
As Polivy is given under the supervision of your doctor it is unlikely that you will be given too much. However, if you experience any side effects after being given Polivy, tell your doctor or nurse immediately.
While you are using Polivy
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Polivy.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
Tell your doctor if you become pregnant or intend to start a family while receiving Polivy.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Things you must not do
Do not stop your Polivy treatment without talking to your doctor first.
Do not take any other medicines, whether they require a prescription or not, without first telling your doctor or consulting with a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how Polivy affects you. Polivy may cause slight light-headedness, tiredness and dizziness in some people. If you experience infusion-related reactions or nerve damage, or if you feel tired, weak or dizzy, do not drive or use machines until symptoms stop.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Polivy.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Because Polivy is given with other medicines to treat cancer it may be difficult for your doctor to tell whether the side effects are due to Polivy or due to other medicines.
Ask your doctor or pharmacist to answer any questions you may have.
During an infusion
Tell your doctor or nurse immediately if you notice any of the following while receiving an infusion (particularly during the first infusion):
- swelling of your face, lips, tongue or throat with difficulty breathing
- swelling of other parts of your body such as your hands or feet
- shortness of breath, wheezing or trouble breathing
- abnormal or irregular heartbeat
- rash, itching or hives on the skin
- flushing (warm, red) skin
- pain or swelling at site of injection
- feeling sick (nausea) or vomiting, diarrhoea
- pain or discomfort (including stomach pain, back pain, chest or neck pain)
- fever or chills
After an infusion
Tell your doctor straight away if you notice any of the side effects below or if they get worse. They may happen weeks or months after your last dose. Do not try to treat yourself with other medicines.
- Flu and/or cold-like symptoms, chest pain, coughing, sweating
- Fever, sore throat, tiredness, sores in the mouth or gums
- Bruising, bleeding gums or nose, rash on legs with red dots, blood in urine or stools
- Numbness or tingling in the hands or feet, sharp or jabbing pain, burning or freezing sensation, pins and needles
- Weakness, lack of energy, feeling unsteady
- Muscle cramps or spasms, heart palpitations, breathing difficulties, mood changes
- Swelling of the hands or feet, yellow skin or eyes, rapid heartbeat, appetite changes
- Confusion or memory loss, muscle spasms and cramps, facial twitching, numbness
- Nose bleeds, feeling dizzy, tired, looking pale
- Nausea or vomiting
- Constipation or abdominal pain
- Diarrhoea
- Rash, itching or hives on the skin
- Decreased appetite, weight decrease
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
Some side effects can only be found when your doctor does tests from time to time to check your progress. (for example, elevated liver enzymes, low potassium or sodium levels, low platelet count, low blood pressure).
Product description
Storage
Polivy will be stored in the pharmacy or on the hospital ward in a refrigerator at a temperature between 2°C and 8°C. Polivy should not be frozen.
Availability
Polivy is supplied as a single-dose glass vial containing 140 mg of active ingredient.
It is diluted before infusion into a vein.
What it looks like
Polivy is a white to grey powder which is dissolved in sterile water before use.
Ingredients
Polivy contains 140 mg of polatuzumab vedotin as the active ingredient.
It also contains:
- succinic acid
- sodium hydroxide
- sucrose
- polysorbate 20
This medicine does not contain lactose, gluten, tartrazine or any other azo dyes.
Manufacturer
Polivy is distributed in Australia by:
Roche Products Pty Limited
ABN 70 000 132 865
Level 8, 30 - 34 Hickson Road
Sydney NSW 2000
AUSTRALIA
Customer enquiries: 1800 233 950
Please check with your pharmacist for the latest Consumer Medicine Information.
Australian Registration Numbers
- AUST R 314866
This leaflet was prepared on 13 May 2021.
Published by MIMS August 2021
 For dose modifications to manage myelosuppression, see Table 2.
For dose modifications to manage myelosuppression, see Table 2. For dose modifications to manage infusion-related reactions, see Table 3.
For dose modifications to manage infusion-related reactions, see Table 3.
 3. Withdraw the required volume of reconstituted solution from the Polivy vial using a sterile syringe and dilute into the IV infusion bag. Discard any unused portion left in the vial.
3. Withdraw the required volume of reconstituted solution from the Polivy vial using a sterile syringe and dilute into the IV infusion bag. Discard any unused portion left in the vial. Avoid transportation of the prepared solution for infusion as agitation stress can result in aggregation. If the prepared solution for infusion will be transported, remove air from the infusion bag and limit transportation to 30 minutes at 9°C to 25°C or 12 hours at 2°C to 8°C. If air is removed, an infusion set with a vented spike is required to ensure accurate dosing during the infusion. The total storage plus transportation times of the diluted product should not exceed the storage duration specified in Table 4.
Avoid transportation of the prepared solution for infusion as agitation stress can result in aggregation. If the prepared solution for infusion will be transported, remove air from the infusion bag and limit transportation to 30 minutes at 9°C to 25°C or 12 hours at 2°C to 8°C. If air is removed, an infusion set with a vented spike is required to ensure accurate dosing during the infusion. The total storage plus transportation times of the diluted product should not exceed the storage duration specified in Table 4.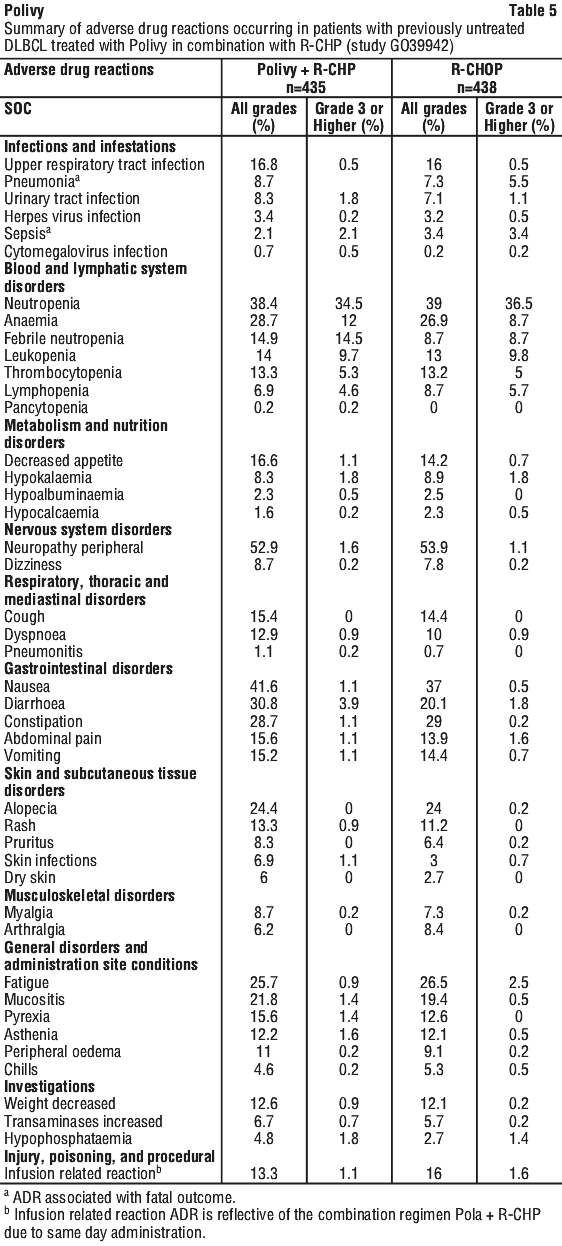


 In the final analysis, overall survival between the Polivy plus R-CHP and R-CHOP arms was similar, HR 0.94 (95% CI: 0.67, 1.33).
In the final analysis, overall survival between the Polivy plus R-CHP and R-CHOP arms was similar, HR 0.94 (95% CI: 0.67, 1.33). In the Polivy plus BR arm, of the 25 patients who achieved a partial or complete response, 16 (64%) had a DOR of at least 6 months, and 12 (48%) had a DOR of at least 12 months. In the BR arm, of the 10 patients who achieved a partial or complete response, 3 (30%) had a DOR lasting at least 6 months, and 2 (20%) had a DOR lasting at least 12 months.
In the Polivy plus BR arm, of the 25 patients who achieved a partial or complete response, 16 (64%) had a DOR of at least 6 months, and 12 (48%) had a DOR of at least 12 months. In the BR arm, of the 10 patients who achieved a partial or complete response, 3 (30%) had a DOR lasting at least 6 months, and 2 (20%) had a DOR lasting at least 12 months.