

What is in this leaflet
This leaflet answers some common questions about Pradaxa.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the expected benefits.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
This leaflet was last updated on the date at the end of this leaflet. More recent information may be available. The latest Consumer Medicine Information is available from your pharmacist, doctor or from www.medicines.org.au and may contain important information about the medicine and its use of which you should be aware.
Keep this information with the medicine. You may need to read it again.
What Pradaxa is used for
Pradaxa contains the active substance dabigatran etexilate (as dabigatran etexilate mesilate). After oral use, dabigatran etexilate is rapidly converted in the body to its active form dabigatran. It belongs to a group of medicines called anticoagulants. Some people refer to anticoagulant medicines as "blood thinners". Dabigatran works by inhibiting a specific protein in the blood, called thrombin. Thrombin contributes to the formation of blood clots. Dabigatran prevents the formation of blood clots.
Pradaxa has been prescribed to you for one of the following uses:
- to prevent the formation of blood clots in the veins after knee or hip replacement surgery in adults
- to reduce the risk of brain (stroke) and/or other body vessel obstruction by blood clot formation in adults with an abnormal heart beat rhythm called non-valvular atrial fibrillation
- to treat blood clots in the veins of your legs and lungs and to prevent blood clots from re-occurring in the veins of your legs and/or lungs.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
Before you take Pradaxa
When you must not take it
Do not take Pradaxa if you are allergic to:
- dabigatran etexilate or any of the other ingredients in Pradaxa listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- rash, itching or hives on the skin
- swelling of the face, lips, tongue or other parts of the body
- shortness of breath, wheezing or troubled breathing.
Do not take Pradaxa:
- if you are currently bleeding
- if you have severely reduced kidney function (your doctor will know how to determine your kidney function)
- if you have an increased tendency of bleeding complications (this may be inherited, of unknown cause or due to other medicines)
- if you have a medical condition which increases your risk of serious bleeding, such as recent brain or spinal injury, and cancer
- if you have active stomach ulcers or have experienced stomach bleeding in the past year, unless the cause has been permanently eliminated, e.g. by surgery
- if you have a history of bleeding in the head, eyes, spine, abdomen and joints
- if you have an indwelling spinal or epidural catheter, and during the first two hours after their removal (your doctor will know about the kind of catheters and precautionary measures)
- if you have liver problems or liver disease
- if you are currently taking oral ketoconazole or itraconazole, medicines used to treat fungal infections
- if you are taking dronedarone, a medicine used to treat abnormal heart beat
- if you are taking ciclosporin or tacrolimus, medicines used to prevent organ rejection after transplantation
- if you are taking glecaprevir/pibrentasvir, a combination medicine used to treat hepatitis C infection
- if you are taking medicines to prevent blood clotting (e.g. warfarin, rivaroxaban, apixaban or heparin), except when changing anticoagulant treatment, while having a venous or arterial line and you get heparin through this line to keep it open or while your heart beat is being restored to normal by a procedure called catheter ablation for atrial fibrillation
- if you have a prosthetic heart valve.
Do not start Pradaxa and verapamil treatment at the same time.
Do not start verapamil if you are currently taking Pradaxa and have just undergone major orthopaedic surgery.
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breastfeed if you are taking this medicine. The active ingredient in Pradaxa passes into breast milk.
Do not give this medicine to a child or adolescent under 18 years old.
Do not take this medicine after the expiry date printed on the box or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal. If you use this medicine after the expiry date has passed, it may not work as well.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have any allergies to any other medicines or any other substances, such as foods, preservatives or dyes.
Tell your doctor if you are pregnant or intend to become pregnant. Pradaxa should not be used during pregnancy.
Tell your doctor if you are breastfeeding or intend to breastfeed. Pradaxa is not recommended in women who are breastfeeding.
Tell your doctor if you have had a heart attack or if you have been diagnosed with conditions that increase the risk to develop a heart attack.
Tell your doctor if you have reduced liver function, life-threatening liver disease or increased liver enzymes.
Tell your doctor if you have an increased bleeding risk, as could be the case in the following situations:
- if you are older than 75 years, your doctor may prescribe a lower dose of Pradaxa
- if you know you have reduced kidney function, or you are suffering from dehydration (symptoms include feeling thirsty and passing reduced amounts of dark-coloured urine)
- if you have been recently bleeding
- if you have any problems with your blood
- if you have had a recent tissue sampling (biopsy)
- if you have cancer
- if you have had a serious injury (e.g. a bone fracture, head injury or any injury requiring treatment)
- if you are suffering from an inflammation of the food pipe (oesophagus) or stomach
- if you have problems with reflux of gastric juice into the food pipe (oesophagus)
- if you are receiving medicines which could increase the risk of bleeding, such as clopidogrel and warfarin
- if you are taking anti-inflammatory medicines such as diclofenac
- if you are suffering from an infection of the heart (bacterial endocarditis).
Tell your doctor if you know that you have a disease called antiphospholipid syndrome (a disorder of the immune system that causes an increased risk of blood clots).
If you have not told your doctor about any of the above, tell them before you use Pradaxa.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Pradaxa may interfere with each other. These include:
- aspirin, salicylates or other NSAID (anti-inflammatory) medicines
- medicines used to thin your blood (such as warfarin, unfractionated heparins, heparin derivatives (fondaparinux and desirudin), low molecular weight heparins (enoxaparin), clopidogrel, tirofiban, bivalirudin, prasugrel, ticagrelor, eptifibatide, ticlopidine, dextran, sulfinpyrazone, rivaroxaban and apixaban)
- amiodarone, dronedarone, medicines used to treat irregular heartbeats
- verapamil, a calcium channel blocker used to treat high blood pressure and angina
- quinidine, a medicine used to treat malaria and irregular heartbeats
- clarithromycin or rifampicin, medicines used to treat infections
- lopinavir, nelfinavir, ritonavir, tipranavir or saquinavir, medicines used to treat HIV infections
- ciclosporin or tacrolimus, medicines used to help the body's immune system
- glecaprevir/pibrentasvir, a combination medicine used to treat hepatitis C infection
- selective serotonin re-uptake inhibitors (SSRI) (e.g. citalopram, escitalopram, fluoxetine), selective serotonin norepinephrine re-uptake inhibitors (SNRI) (e.g. duloxetine, venlafaxine, desvenlafaxine), medicines used to treat mood disorders
- herbal medicines derived from St John's wort (Hypericum perforatum)
- carbamazepine, a medicine used to treat fits or convulsions
- medicines used to treat reflux and stomach ulcers (such as pantoprazole and ranitidine).
These medicines may be affected by Pradaxa or may affect how well it works. You may need different amounts of your medicines, change the timing of your medicine-taking routine or take different medicines. Your doctor or pharmacist will advise you.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking Pradaxa.
How to take Pradaxa
Follow the instructions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
AFTER KNEE AND HIP REPLACEMENT SURGERY
The recommended dose of Pradaxa is 220 mg (2 capsules of 110 mg) taken as a single dose once daily.
Patients with moderately reduced kidney function (e.g. over 75 years) or patients taking certain medicines may have an increased risk of bleeding. The doctor may prescribe the lower dose of 150 mg once daily, taken as 2 capsules of Pradaxa 75 mg.
Treatment with Pradaxa should be started within 1 - 4 hours of completed surgery, using a single capsule of 110 mg and continuing with 2 capsules of 110 mg once daily for a total of 10 days (after knee replacement surgery) or for a total of 28 - 35 days (after hip replacement surgery).
If, within 4 hours after surgery, post-operative bleedings can still be observed, initiation of treatment should be delayed. If treatment is not started on the day of surgery then treatment should be initiated with 2 capsules of 110 mg once daily.
Follow the initiation instructions given to you by your doctor carefully.
FOR STROKE PREVENTION IN PATIENTS WITH ATRIAL FIBRILLATION
The recommended dose of Pradaxa is 300 mg taken as 1 capsule of 150 mg in the morning and 1 capsule of 150 mg in the evening.
Patients over 75 years should take a lower dose of 220 mg, taken as 1 capsule of 110 mg in the morning and 1 capsule of 110 mg in the evening.
Patients with an increased risk of major bleeding (as determined by your doctor) should take a lower dose of 220 mg, taken as 1 capsule of 110 mg in the morning and 1 capsule of 110 mg in the evening.
FOR THE TREATMENT OF BLOOD CLOTS AND PREVENTION OF BLOOD CLOTS RE-OCCURRING IN THE VEINS OF YOUR LEGS AND/OR LUNGS
The recommended dose of Pradaxa is 300 mg taken as 1 capsule of 150 mg twice a day following treatment with an injectable blood thinner for at least 5 days. To prevent blood clots re-occurring, continue on 1 capsule of 150 mg twice a day.
Patients over 75 years should take a lower dose of 220 mg, taken as 1 capsule of 110 mg in the morning and 1 capsule of 110 mg in the evening. To prevent blood clots re-occurring, continue on 1 capsule of 110 mg twice a day.
Patients with an increased risk of major bleeding (as determined by your doctor) should take a lower dose of 220 mg, taken as 1 capsule of 110 mg in the morning and 1 capsule of 110 mg in the evening. To prevent blood clots re-occurring, continue on 1 capsule of 110 mg twice a day.
Your doctor will decide how long you need to be on this treatment for.
How to take it
Pradaxa is available in blister packs.
REMOVING PRADAXA CAPSULES FROM THE BLISTER PACK
Prior to removing a capsule from the blister card, separate one blister segment by tearing along the perforations (Figure A).
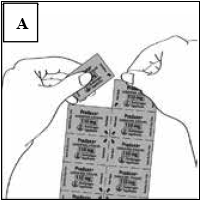
Once you have separated an individual blister segment, locate the tab marked with the arrow (Figure B).
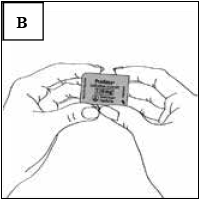
Immediately before you are ready to take your dose of Pradaxa, peel back the foil using the tab marked with the arrow until the capsule is fully visible (Figure C).
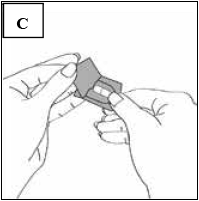
Turn the blister segment upside down and tip the capsule out, tapping the back of the blister segment, if necessary.
Do not try to push the capsule through an unopened blister segment.
Do not cut the foil or use sharp instruments to remove the capsule from the blister.
Capsules should always be stored in the sealed blister segments and only removed immediately before use. The capsule should be taken immediately after the foil over an individual blister segment is opened, or its effectiveness may be reduced.
If additional capsules are inadvertently exposed to air, they should not be used and should be discarded.
Capsules should not be removed from the blister pack and repackaged in dose administration aids such as dosette boxes, tablet organisers or weekly medication packs.
Swallow the capsules whole with a full glass of water.
DO NOT CHEW OR OPEN THE CAPSULE. DO NOT SPRINKLE THE PELLETS ON FOOD OR MIX WITH LIQUIDS. This may cause an overdose of Pradaxa and increase the risk of bleeding.
When to take it
Take Pradaxa at about the same time each day. Taking your capsules at the same time each day will have the best effect. It will also help you remember when to take it. It does not matter if you take this medicine with or without food.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
Pradaxa will continue to be prescribed while there is a risk of excessive clotting.
AFTER KNEE REPLACEMENT SURGERY:
This will usually be for a period of 10 days.
AFTER HIP REPLACEMENT SURGERY:
This will usually be for a period of 28 - 35 days.
It is important to keep taking your medicine even if you feel well. If you stop using Pradaxa before your doctor tells you to stop, you are at risk of developing a blood clot in a vein of your leg which can move to the lungs and be life-threatening.
Tell your doctor immediately or go to Emergency at your nearest hospital if you notice swelling of the leg or cough and shortness of breath. These could be signs of a blood clot.
Tell your doctor if you intend stopping treatment earlier.
FOR STROKE PREVENTION IN PATIENTS WITH ATRIAL FIBRILLATION:
It is important to keep taking your medicine even if you feel well. If you stop using Pradaxa before your doctor tells you to stop, you are at risk of developing a blood clot. This can lead to serious health problems such as strokes.
FOR TREATMENT AND PREVENTION OF BLOOD CLOTS RE-OCCURRING IN THE VEINS OF YOUR LEGS AND LUNGS:
It is important to keep taking your medicine even if you feel well. If you stop using Pradaxa before your doctor tells you to stop, you are at risk of developing a blood clot. This can lead to serious health problems if those clots stop blood flowing normally.
If you forget to take it
After knee and hip replacement surgery continue with your remaining daily doses of Pradaxa at the same time of the next day.
Do not take a double dose to make up for missed individual doses.
For stroke prevention in patients with atrial fibrillation a forgotten dose of Pradaxa can still be taken up to 6 hours prior to the next dose.
A missed dose should be omitted if the remaining time is less than 6 hours prior to the next dose.
Do not take a double dose to make up for missed individual doses.
For the treatment and prevention of blood clots re-occurring in the veins of your legs and lungs a forgotten dose of Pradaxa can still be taken up to 6 hours prior to the next dose.
A missed dose should be omitted if the remaining time is less than 6 hours prior to the next dose. Do not take a double dose to make up for missed individual doses.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for hints.
If you take too much (Overdose)
Immediately telephone your doctor or Poisons Information Centre (Australia 13 11 26) for advice, or go to Emergency at your nearest hospital if you think that you or anyone else may have taken too much Pradaxa.
Do this even if there are no signs of discomfort or poisoning.
If you take too much Pradaxa you may have bleeding. Blood may be seen in stools or urine. Abnormal bruising may also be experienced.
While you are taking Pradaxa
Things you must do
Tell all doctors and pharmacists who are treating you that you are taking Pradaxa.
Tell your doctor if, for any reason, you have not used Pradaxa exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
If you become pregnant while using Pradaxa, tell your doctor immediately.
If you are going to have surgery, including dental surgery, tell your doctor or dentist that you are taking Pradaxa.
Your doctor may decide to temporarily stop your treatment with Pradaxa.
Tell your doctor if you fall or injure yourself during treatment, especially if you hit your head, please seek urgent medical attention. You may need to be checked by a doctor, as you may be at increased risk of bleeding.
Things you must not do
Do not give Pradaxa to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
No studies on the effects of Pradaxa on the ability to drive and operate machinery have been performed.
Driving or operating machinery should be avoided for a period of time after orthopaedic surgery.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Pradaxa.
All medicines can have side effects. Sometimes they are serious, most of the time they are not.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- bruising
- nose bleeds
- stomach ache
- itchy skin, rash
- diarrhoea
- indigestion
- feeling sick
- cough
- painful, swollen joints
- sore nasal passages and throat
- discomfort when swallowing
- hair loss
- frequent infections such as fever, severe chills, sore throat or mouth ulcers (signs of lack of white blood cells).
These side effects are usually mild.
Tell your doctor immediately or go to Emergency at your nearest hospital if you notice any of the following:
- long or excessive bleeding
- exceptional weakness
- tiredness, headaches, dizziness and looking pale (signs of anaemia)
- chest pain or being short of breath
- swelling of hands, ankles and feet
- red or dark brown urine
- red or black bowel motions
- symptoms related to a condition called anticoagulant-related nephropathy: blood in urine, reduced urine output, swelling of the legs, ankles and feet, increased time for blood to clot (high INR test values), heavy bleeding.
These are serious side effects. You may need urgent medical attention.
Other side effects not listed above may occur in some people.
Tell your doctor if you notice anything else that is making you feel unwell.
After using Pradaxa
Storage
Keep your capsules in the blister pack until it is time to take them. If you take them out of the blister pack they may not keep well.
Keep Pradaxa in a cool dry place where the temperature stays below 30°C.
Do not store Pradaxa or any other medicine in the bathroom or near a sink.
Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines.
Keep Pradaxa where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking Pradaxa or it has passed its expiry date, ask your pharmacist what to do with any that is left over.
Product Description
What it looks like
Pradaxa is the brand name of your medicine.
Pradaxa is available in three strengths of capsules:
- Pradaxa 75 mg - white-coloured, opaque cap and body, imprinted with a R75 code on one side and company logo on the other
- Pradaxa 110 mg - light blue-coloured, opaque cap and body, imprinted with a R110 code on one side and company logo on the other
- Pradaxa 150 mg - light blue-coloured, opaque cap with a white-coloured, opaque body imprinted with a R150 code on one side and company logo on the other.
Pradaxa 75 mg, 110 mg and 150 mg are available in blister packs of 10 and 60 capsules. Pradaxa 75 mg and 110 mg are also available in blister packs of 30* capsules.
* Not distributed in Australia.
Ingredients
Active ingredient:
- Pradaxa 75 mg - 75 mg dabigatran etexilate given as 86.48 mg dabigatran etexilate mesilate per capsule.
- Pradaxa 110 mg - 110 mg dabigatran etexilate given as 126.83 mg dabigatran etexilate mesilate per capsule.
- Pradaxa 150 mg - 150 mg dabigatran etexilate given as 172.95 mg dabigatran etexilate mesilate per capsule.
Inactive ingredients:
Capsule fill
- acacia
- dimeticone 350
- hyprolose
- hypromellose
- purified talc
- tartaric acid
Capsule shell
- carrageenan
- potassium chloride
- titanium dioxide
- indigo carmine CI73015 (110 mg and 150 mg capsules only)
- hypromellose
- purified water
Black printing ink
- TekPrint SW-9008 Black Ink.
Pradaxa does not contain gluten, sucrose or tartrazine.
Supplier
Pradaxa is supplied in Australia by:
Boehringer Ingelheim Pty Limited
ABN 52 000 452 308
Sydney, Australia
www.boehringer-ingelheim.com.au
This Consumer Medicine Information was updated in May 2023
® Pradaxa is a registered trademark of Boehringer Ingelheim.
© Boehringer Ingelheim Pty Limited 2023
Australian Registration Numbers
Pradaxa 75 mg: AUST R 137832
Pradaxa 110 mg: AUST R 138402
Pradaxa 150 mg: AUST R 168211
Published by MIMS June 2023
 This method is recommended when assessing patients' CrCL prior to and during dabigatran treatment.
This method is recommended when assessing patients' CrCL prior to and during dabigatran treatment. See Table 1.
See Table 1.
 In atrial fibrillation patients in RE-LY treated with 150 mg twice daily, an aPTT of greater than 2.0-3.0 fold of normal range at trough was associated with an increased risk of bleeding.
In atrial fibrillation patients in RE-LY treated with 150 mg twice daily, an aPTT of greater than 2.0-3.0 fold of normal range at trough was associated with an increased risk of bleeding. Patients ≥ 75 years of age should not be treated with Pradaxa 150 mg twice a day (see Section 4.2 Dose and Method of Administration, Prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation).
Patients ≥ 75 years of age should not be treated with Pradaxa 150 mg twice a day (see Section 4.2 Dose and Method of Administration, Prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation). Patients taking dabigatran etexilate with PPIs or H2-blockers may be at increased risk of gastrointestinal bleeding due to the associated gastrointestinal conditions for which these drugs are prescribed.
Patients taking dabigatran etexilate with PPIs or H2-blockers may be at increased risk of gastrointestinal bleeding due to the associated gastrointestinal conditions for which these drugs are prescribed. Dabigatran etexilate is contraindicated in patients with severe renal dysfunction (CrCL < 30 mL/min) but should this occur then dabigatran etexilate should be stopped at least 5 days before major surgery.
Dabigatran etexilate is contraindicated in patients with severe renal dysfunction (CrCL < 30 mL/min) but should this occur then dabigatran etexilate should be stopped at least 5 days before major surgery. Overall bleeding rates were similar between treatment groups and not significantly different.
Overall bleeding rates were similar between treatment groups and not significantly different. Adverse reactions observed with exposure to dabigatran etexilate 150 mg daily and 220 mg daily from all 6 controlled VTE prevention studies are listed below by system organ class and frequency according to the following categories: common ≥ 1% and < 10%, uncommon ≥ 0.1% and < 1%, rare ≥ 0.01% and < 0.1%.
Adverse reactions observed with exposure to dabigatran etexilate 150 mg daily and 220 mg daily from all 6 controlled VTE prevention studies are listed below by system organ class and frequency according to the following categories: common ≥ 1% and < 10%, uncommon ≥ 0.1% and < 1%, rare ≥ 0.01% and < 0.1%. The pattern of adverse events for RE-NOVATE II (1160.64) was similar to RE-NOVATE (1160.48).
The pattern of adverse events for RE-NOVATE II (1160.64) was similar to RE-NOVATE (1160.48).
 The risk of major bleeding with dabigatran etexilate 110 mg and 150 mg was consistent across all major subgroups of baseline characteristics with the exception of age. There was a higher risk of bleeding with dabigatran etexilate 150 mg in patients ≥ 75 years of age (hazard ratio vs. warfarin (95% CI) 1.19 (0.99, 1.43)).
The risk of major bleeding with dabigatran etexilate 110 mg and 150 mg was consistent across all major subgroups of baseline characteristics with the exception of age. There was a higher risk of bleeding with dabigatran etexilate 150 mg in patients ≥ 75 years of age (hazard ratio vs. warfarin (95% CI) 1.19 (0.99, 1.43)).

 Adverse reactions observed with exposure to dabigatran 110 mg twice daily and 150 mg twice daily during the RE-LY trial are listed below by system organ class and frequency according to the following categories: common ≥ 1% and < 10%, uncommon ≥ 0.1% and < 1%, rare ≥ 0.01% and < 0.1%.
Adverse reactions observed with exposure to dabigatran 110 mg twice daily and 150 mg twice daily during the RE-LY trial are listed below by system organ class and frequency according to the following categories: common ≥ 1% and < 10%, uncommon ≥ 0.1% and < 1%, rare ≥ 0.01% and < 0.1%. Bleeding events for both treatments are counted from the first intake of dabigatran etexilate or warfarin after the parenteral therapy has been discontinued (oral only treatment period). This includes all bleeding events which occurred during dabigatran therapy. All bleeding events which occurred during warfarin therapy are included except for those during the overlap period between warfarin and parenteral therapy.
Bleeding events for both treatments are counted from the first intake of dabigatran etexilate or warfarin after the parenteral therapy has been discontinued (oral only treatment period). This includes all bleeding events which occurred during dabigatran therapy. All bleeding events which occurred during warfarin therapy are included except for those during the overlap period between warfarin and parenteral therapy. The definition of MBEs followed the recommendations of the International Society on Thrombosis and Haemostasis as described under RE-COVER and RE-COVER II.
The definition of MBEs followed the recommendations of the International Society on Thrombosis and Haemostasis as described under RE-COVER and RE-COVER II.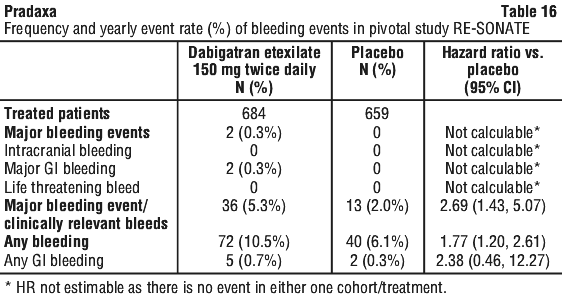 The definition of MBEs followed the recommendations of the International Society on Thrombosis and Haemostasis as described under RE-COVER and RE-COVER II.
The definition of MBEs followed the recommendations of the International Society on Thrombosis and Haemostasis as described under RE-COVER and RE-COVER II.

 The overall frequency of adverse reactions in patients receiving Pradaxa for acute DVT/PE treatment was lower for Pradaxa compared to warfarin (14.2% vs. 18.9%).
The overall frequency of adverse reactions in patients receiving Pradaxa for acute DVT/PE treatment was lower for Pradaxa compared to warfarin (14.2% vs. 18.9%). The overall frequency of adverse reactions in patients treated for recurrent DVT/PE prevention was lower for Pradaxa compared to warfarin (14.6% vs. 19.6%); compared to placebo the frequency was higher (14.6% vs. 6.5%).
The overall frequency of adverse reactions in patients treated for recurrent DVT/PE prevention was lower for Pradaxa compared to warfarin (14.6% vs. 19.6%); compared to placebo the frequency was higher (14.6% vs. 6.5%). Table 22 presents the combined incidences of major VTE and VTE related mortality for RE-MODEL and RE-NOVATE trials. The most frequent component of the composite endpoint was proximal DVT in all three treatment groups. Non-fatal pulmonary embolism (PE) during the treatment period in the two trials were observed in 1 patient in the dabigatran etexilate 150 mg group, 3 patients receiving enoxaparin and 5 patients receiving dabigatran etexilate 220 mg. VTE related mortality was observed for 1 patient in each of the dabigatran etexilate 220 mg and enoxaparin groups and for 4 patients in the dabigatran etexilate 150 mg group. See Table 23.
Table 22 presents the combined incidences of major VTE and VTE related mortality for RE-MODEL and RE-NOVATE trials. The most frequent component of the composite endpoint was proximal DVT in all three treatment groups. Non-fatal pulmonary embolism (PE) during the treatment period in the two trials were observed in 1 patient in the dabigatran etexilate 150 mg group, 3 patients receiving enoxaparin and 5 patients receiving dabigatran etexilate 220 mg. VTE related mortality was observed for 1 patient in each of the dabigatran etexilate 220 mg and enoxaparin groups and for 4 patients in the dabigatran etexilate 150 mg group. See Table 23.





 The RE-LY extension study (RELY-ABLE) provided additional safety information for a large cohort of patients which continued the same dose of dabigatran etexilate as assigned in the RE-LY trial. Patients were eligible for the RELY-ABLE trial if they had not permanently discontinued study medication at the time of their final RE-LY study visit. Enrolled patients continued to receive the same double-blind dabigatran etexilate dose randomly allocated in RE-LY, for up to 43 months of follow-up after RE-LY (total mean follow-up RE-LY + RELY-ABLE, 4.5 years). There were 5897 patients enrolled, representing 49% of patients originally randomly assigned to receive dabigatran etexilate in RE-LY and 86% of RELY-ABLE-eligible patients.
The RE-LY extension study (RELY-ABLE) provided additional safety information for a large cohort of patients which continued the same dose of dabigatran etexilate as assigned in the RE-LY trial. Patients were eligible for the RELY-ABLE trial if they had not permanently discontinued study medication at the time of their final RE-LY study visit. Enrolled patients continued to receive the same double-blind dabigatran etexilate dose randomly allocated in RE-LY, for up to 43 months of follow-up after RE-LY (total mean follow-up RE-LY + RELY-ABLE, 4.5 years). There were 5897 patients enrolled, representing 49% of patients originally randomly assigned to receive dabigatran etexilate in RE-LY and 86% of RELY-ABLE-eligible patients.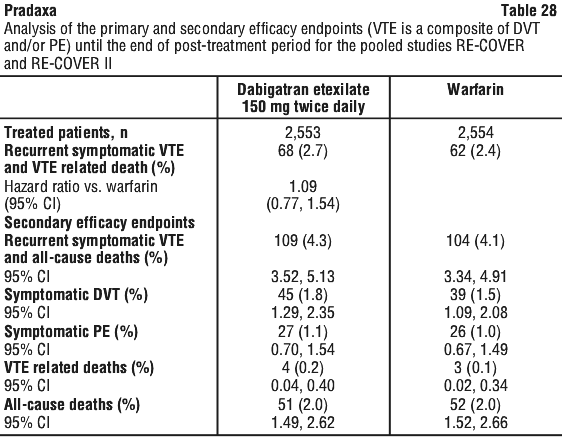
 The objective of the RE-SONATE study was to evaluate superiority of dabigatran etexilate versus placebo for the prevention of recurrent symptomatic DVT and/or PE in patients who had already completed 6 to 18 months of treatment with VKA. The intended therapy was 6 months dabigatran etexilate 150 mg twice daily without need for monitoring.
The objective of the RE-SONATE study was to evaluate superiority of dabigatran etexilate versus placebo for the prevention of recurrent symptomatic DVT and/or PE in patients who had already completed 6 to 18 months of treatment with VKA. The intended therapy was 6 months dabigatran etexilate 150 mg twice daily without need for monitoring.
 Upon administration of the dabigatran etexilate HPMC capsules together with a high fat, high caloric breakfast, the average total exposure (AUC) of dabigatran increased by 27% and the maximum exposure on average by 8.5%. The time to peak plasma concentrations was delayed by 2 hours. The relative increase of bioavailability was considered of no clinical relevance.
Upon administration of the dabigatran etexilate HPMC capsules together with a high fat, high caloric breakfast, the average total exposure (AUC) of dabigatran increased by 27% and the maximum exposure on average by 8.5%. The time to peak plasma concentrations was delayed by 2 hours. The relative increase of bioavailability was considered of no clinical relevance. Similar findings were observed in the RE-LY study. The median CrCL in RE-LY was 68.4 mL/min. Almost half (45.8%) of the RE-LY patients had a CrCL between 50-80 mL/min. When compared with patients without renal impairment (CrCL ≥ 80 mL/min), patients with moderate renal impairment (CrCL between 30-50 mL/min) had pre- and post-dose dabigatran plasma concentrations 2.29-fold and 1.81-fold higher on average, respectively.
Similar findings were observed in the RE-LY study. The median CrCL in RE-LY was 68.4 mL/min. Almost half (45.8%) of the RE-LY patients had a CrCL between 50-80 mL/min. When compared with patients without renal impairment (CrCL ≥ 80 mL/min), patients with moderate renal impairment (CrCL between 30-50 mL/min) had pre- and post-dose dabigatran plasma concentrations 2.29-fold and 1.81-fold higher on average, respectively. Molecular Formula: C35H45N7O8S.
Molecular Formula: C35H45N7O8S.