What is in this leaflet
This leaflet answers some common questions about PREZISTA (pre-ZIS-ta) tablets. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given PREZISTA against the benefits this medicine is expected to have for you.
If you have any concerns about being given PREZISTA ask your doctor.
Keep this leaflet while you are taking PREZISTA. You may need to read it again.
What PREZISTA is used for
PREZISTA is an antiretroviral medicine. It belongs to a group of medicines called protease (PRO-tee-ase) inhibitors. PREZISTA works by reducing the amount of HIV in your body. Reducing the amount of HIV in your blood improves your immune system, and reduces the risk of developing illnesses as a result of HIV infection.
PREZISTA is used to treat adults, and children 6 years of age or above weighing more than 20 kg, who are infected by HIV (Human Immunodeficiency Virus).
In adults, PREZISTA must be taken in combination with cobicistat or with a low dose of ritonavir, and with other anti-HIV medicines.
In children, PREZISTA must be taken in combination with a low dose of ritonavir, and with other anti-HIV medicines.
Your doctor will discuss with you which combination of medicines will work best with PREZISTA.
Ask your doctor if you have any questions about why PREZISTA has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you take PREZISTA
When you must not use it:
Do not take PREZISTA:
- if you are allergic (hypersensitive) to darunavir, other ingredients of PREZISTA (listed in Ingredients section) or to ritonavir or to cobicistat.
Symptoms of an allergic reaction may include rash, itching or hives on the skin, shortness of breath, wheezing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body.
Do not take PREZISTA:
- if the packaging is torn or shows signs of tampering.
- if the expiry date (month and year) printed on the pack has passed. If you take PREZISTA after the expiry date it may not work.
PREZISTA should not be given to children younger than 6 years of age. PREZISTA should not be given to children and adolescents who have never used antiretroviral medicines before.
Do not take PREZISTA in combination with ritonavir or cobicistat with any of the following medicines:
- amiodarone, bepridil, disopyramide, flecainide, mexiletine, propafenone, lidocaine, quinidine or dronedarone (to treat irregular heartbeats)
- ivabradine or ranolazine (to treat heart disease)
- midazolam or triazolam (to treat trouble with sleeping and/or anxiety)
- ergot alkaloids i.e. dihydroergotamine, ergonovine, ergotamine, methylergonovine (to treat migraine and headaches)
- astemizole or terfenadine (to treat allergy symptoms)
- cisapride (to treat some stomach conditions)
- lurasidone or pimozide (to treat psychiatric conditions)
- alfuzosin (to treat an enlarged prostate)
- sildenafil (to treat pulmonary arterial hypertension)
- colchicine (to treat gout) if you have renal/hepatic impairment
- lovastatin, simvastatin or lomitapide (to lower cholesterol levels)
- apixaban (used to reduce blood clotting)
- rifampicin (to treat some infections such as tuberculosis)
- elbasvir/grazoprevir (to treat hepatitis C)
- products that contain St John's wort (Hypericum perforatum)
- naloxegol (to treat opioid induced constipation)
- dapoxetine (to treat premature ejaculation)
Do not take PREZISTA in combination with cobicistat with:
certain medicines to treat epilepsy and prevent seizures (carbamazepine, phenobarbital or phenytoin). If you are taking any of these, ask your doctor about switching to another medicine.
This is not a complete list of medicines. Therefore, tell your doctor about all medicines you take.
There are other medicines that you need to be careful of when taking PREZISTA (see Taking other medicines).
Before you start to use it:
Take special care with PREZISTA:
PREZISTA is not a cure for HIV infection.
PREZISTA does not reduce the risk of passing HIV to others through sexual contact or blood. Therefore, you must continue to use appropriate precautions to prevent passing HIV on to others.
People taking PREZISTA can still develop infections or other illnesses associated with HIV. You should continue to keep in regular contact with your doctor and to monitor your health while taking PREZISTA.
Tell your doctor if you have or have had any medical conditions, especially the following:
- Problems with your liver, including hepatitis B and C.
Your doctor may need to evaluate your liver before deciding if you can take PREZISTA. - Diabetes.
PREZISTA, like some other anti-HIV medicines, might increase sugar levels in the blood. - Haemophilia.
Anti-HIV medicines, such as PREZISTA, might increase the risk of bleeding in patients with this blood clotting disorder. - An allergy to sulfa medicines (sulphonamides).
Tell your doctor immediately if you are pregnant or breastfeeding, or intend to become pregnant or breastfeed.
Do not take PREZISTA with cobicistat if you are pregnant or breastfeeding. During pregnancy and breastfeeding you must not take PREZISTA with ritonavir, unless it is specifically approved by your doctor. It is recommended that HIV infected women should not breastfeed their infants because of the possibility of your baby becoming infected with HIV through your breast milk and because of the unknown effects of the medicine on your baby.
If you have not told your doctor about any of the above, tell them before you start treatment with PREZISTA.
Taking other medicines:
Some medicines may affect the levels of PREZISTA or PREZISTA may affect the level of other medicines in the body when they are taken at the same time as PREZISTA. Your doctor might want to do some additional blood tests.
For this reason, tell your doctor about all medicines you take, including medicines you can buy without a prescription from a pharmacy, supermarket or health food shop.
Know the medicines you take. Keep a list of medicines and show it to your doctor and pharmacist when you get a new medicine. Your doctor and your pharmacist can tell you if you can take these medicines with PREZISTA.
- Tell your doctor if you are taking any of the following medicines. You must not take these medicines while taking PREZISTA in combination with ritonavir or cobicistat: alfuzosin, amiodarone, apixaban, astemizole, bepridil, cisapride, colchicine, dapoxetine, disopyramide, dronedarone, elbasvir/grazoprevir, ergot alkaloids (dihydroergotamine, ergonovine, ergotamine, methylergonovine), flecainide, ivabradine, lidocaine, lomitapide, lovastatin, lurasidone, mexiletine, midazolam, naloxegol, pimozide, propafenone, quinidine, ranolazine, rifampicin, sildenafil, simvastatin, products that contain St John's wort (Hypericum perforatum), terfenadine or triazolam.
- Tell your doctor if you are taking the following medicines to treat epilepsy and prevent seizures. You must not take these medicines while you are taking PREZISTA in combination with cobicistat: carbamazepine, phenobarbital or phenytoin.
- Tell your doctor if you take other anti-HIV medicines. PREZISTA can be combined with some other anti-HIV medicines while other combinations are not recommended.
- If you take PREZISTA with some other medicines, the effects of PREZISTA or other medicines might be influenced. The dosage of some medicines may need to be changed. Some combinations are not recommended. Tell your doctor if you take any of the following:
- contraceptives. PREZISTA might reduce the effectiveness of hormonal contraceptives. Therefore, additional or alternative (non-hormonal) methods of contraception are recommended. If you take a contraceptive containing drospirenone your potassium levels might become elevated.
- medicines for heart disease (amlodipine, diltiazem, felodipine, nifedipine, nicardipine, tadalafil, verapamil).
- medicines to treat certain heart disorders (digoxin, carvedilol, metoprolol, timolol, bosentan).
- medicines used to reduce clotting of the blood (e.g. dabigatran etexilate, edoxaban, rivaroxaban, warfarin) or to prevent blood clots (e.g. ticagrelor, clopidogrel)
- medicines to lower cholesterol levels (atorvastatin, pitavastatin, pravastatin, rosuvastatin). The risk of muscle tissue disorder might be increased.
- medicines for your immune system (cyclosporin, everolimus, tacrolimus, sirolimus). Your doctor might want to do some additional tests.
- medicine to treat asthma (salmeterol).
- corticosteroids (betamethasone, budesonide, dexamethasone, fluticasone, mometasone, prednisone, triamcinolone).
- medicines to treat cancer (dasatinib, everolimus, irinotecan, nilotinib, vinblastine, vincristine).
- medicines to treat narcotic dependence (buprenorphine/ naloxone, methadone)
- medicines to treat malaria (artemether/lumefantrine).
- medicines to treat hepatitis C (glecaprevir/pibrentasvir).
- medicines to treat urinary disorders (fesoterodine, solifenacin).
- medicines to treat nausea and vomiting (domperidone).
- medicines to treat fungal infections (clotrimazole, fluconazole, isavuconazole, itraconazole, ketoconazole, posaconazole voriconazole).
- medicines to treat some infections such as tuberculosis (rifapentine, rifabutin).
- medicines against bacterial infections (clarithromycin).
- medicines to treat gout (colchicine). If you have renal/hepatic impairment, do not take colchicine with PREZISTA.
- medicines for erectile dysfunction (avanafil, sildenafil, vardenafil, tadalafil).
- medicines to treat depression and anxiety (paroxetine, sertraline, amitriptyline, desipramine, imipramine, nortriptyline, and trazodone).
- sedatives (buspirone, clorazepate, diazepam, estazolam, flurazepam, zolpidem).
- medicines to treat excessive sleepiness (armodafinil, modafinil).
- medicines to treat psychiatric conditions (perphenazine, quetiapine, risperidone, thioridazine).
- medicines to treat epilepsy, prevent seizures or to treat trigeminal neuralgia (carbamazepine, clonazepam, oxcarbazepine, phenobarbital, phenytoin).
- certain medicines to treat moderate or severe pain (fentanyl, oxycodone, tramadol). The amount of the pain medicine in the body might increase if it is used with PREZISTA. There is an increased risk of serious breathing difficulties with use or abuse of these pain medicines while on treatment with PREZISTA.
This is not a complete list of medicines. Therefore, tell your doctor about all medicines you take.
Please refer to the ritonavir or cobicistat Consumer Medicine Information for information on ritonavir or cobicistat.
Taking PREZISTA
Adults
Always use PREZISTA exactly as your doctor has told you. You must check with your doctor if you are not sure.
Make sure that you always have enough PREZISTA and ritonavir or cobicistat available so that you don't run out. For example, in case you cannot return home, need to travel or stay in a hospital.
How much PREZISTA to take:
Take PREZISTA exactly as directed by your doctor. Your doctor will decide which dose is right for you.
You must take PREZISTA every day and always in combination with 100 milligrams of ritonavir or 150 milligrams of cobicistat, and with food. PREZISTA cannot work properly without ritonavir or cobicistat, and food. You must eat a meal or a snack within 30 minutes prior to taking your PREZISTA and ritonavir or cobicistat. The type of food is not important.
Even if you feel better, do not stop taking PREZISTA without talking to your doctor.
Instructions:
- Take PREZISTA always together with 100 milligrams of ritonavir or 150 milligrams of cobicistat.
- Take PREZISTA with food.
- Swallow the tablets with a drink such as water, milk, or any other nutritional drink.
Take your other HIV medicines used in combination with PREZISTA and ritonavir or cobicistat as recommended by your doctor.
Children 6 years of age and older, weighing at least 20 kg, who have taken any anti-HIV medicines before (your child's doctor will determine this) The doctor will work out the right dose based on the weight of the child. The doctor will inform you exactly on how many PREZISTA tablets and how much ritonavir (capsules or solution) your child should take.
If your child feels better, do not stop administering PREZISTA without talking to the child's doctor.
Instructions:
- Take PREZISTA always together with ritonavir.
- Take PREZISTA with food.
- Swallow the tablets with a drink such as water, milk, or any other nutritional drink.
- Take your other HIV medicines used in combination with PREZISTA and ritonavir as recommended by your doctor.
- If your child cannot tolerate ritonavir oral solution, consult your doctor
Removing the child resistant cap
The plastic bottle comes with a child resistant cap and should be opened as follows:
- Push the plastic screw cap down while turning it counter clockwise.
- Remove the unscrewed cap.
What do I do if I forget to take PREZISTA?
If you forget to take PREZISTA and your dosing regimen is PREZISTA with ritonavir or cobicistat once a day:
If you notice within 12 hours, you must take the tablets immediately. Always take with ritonavir or cobicistat and food. If you notice after 12 hours, then skip the intake and take the next doses as usual. Do not take a double dose to make up for a forgotten dose.
If you forget to take PREZISTA and your dosing regimen is PREZISTA with ritonavir twice a day:
If you notice within 6 hours, you must take the tablet/s immediately. Always take with ritonavir and food. If you notice after 6 hours, then skip the dose and take the next doses as usual. Do not take a double dose to make up for a forgotten dose.
Please refer to your doctor for instructions on missed doses of other HIV medicines used in combination with PREZISTA and ritonavir or cobicistat.
What do I do if I take too much? (overdose):
If you think you or anybody else has taken too much PREZISTA, contact your doctor, pharmacist or the Poisons Information Centre who will advise you what to do.
You can contact the Poisons Information Centre by dialling:
- Australia: 13 11 26
- New Zealand: 0800 POISON or 0800 764 766.
While you are taking PREZISTA
Things you must do:
- Do not stop taking PREZISTA without talking to your doctor first
HIV therapy may increase your sense of well being. Even when you feel better, do not stop taking PREZISTA. Talk to your doctor first.
Be sure to keep all your doctor's appointments so your progress can be checked. Your doctor will want to do some blood, urine and other tests from time to time to check on your progress.
Tell your doctor if you have any medical conditions, especially the following:
- Symptoms of infection.
In some patients with advanced HIV infection and a history of opportunistic infection, signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body's immune response. This improvement enables the body to fight infections that may have been present prior to taking PREZISTA, with no obvious symptoms.
Be sure to follow up your doctor's instructions about other medicines you should take, and other things you should do.
Ask your doctor or pharmacist if you have any questions.
Tell any other doctors and pharmacists who are treating you that you are taking PREZISTA. If you are undergoing anaesthesia, tell your anaesthetist that you are taking PREZISTA.
If you are about to be started on any new medicines, tell your doctor or pharmacist that you are taking PREZISTA.
If you become pregnant while taking PREZISTA, tell your doctor immediately. You must not take PREZISTA with cobicistat if you are pregnant or breastfeeding.
If you have any further questions on the use of this product, ask your doctor.
Things to be careful of
Driving and using machines
Do not operate machines or drive if you feel dizzy after taking PREZISTA.
Side Effects
Like all medicines, PREZISTA can have side effects. Some of these effects may be serious.
Tell your doctor or pharmacist if you do not feel well while you are being treated with PREZISTA.
When treating HIV infection, it is not always easy to identify what side effects are caused by PREZISTA, which are caused by other medicines you are taking, or which are caused by the HIV infection itself.
The most common side effects are:
- nausea, vomiting
- headache
- abdominal pain, diarrhoea
- indigestion
- passing wind
- rash, (see information below) itching or hives on the skin
- weakness or lack of energy
- feeling tired
PREZISTA may change some values of your blood chemistry. These can be seen in the results of blood tests. Your doctor will explain these to you.
Liver problems that may occasionally be severe have been reported. Your doctor should do blood tests prior to initiating PREZISTA. If you have chronic hepatitis B or C infection, your doctor should check your blood tests more often because you have an increased chance of developing liver problems. Talk to your doctor about the signs and symptoms of liver problems. These may include yellowing of your skin or whites of your eyes, dark (tea coloured) urine, pale coloured stools (bowel movements), nausea, vomiting, loss of appetite, or pain, aching, or sensitivity on your right side below your ribs.
Skin rash has been reported in 10% of patients receiving PREZISTA. Occasionally a rash can be severe or potentially life threatening. In patients taking PREZISTA and raltegravir, rashes (generally mild or moderate) may occur more frequently that in patients taking either drug separately. It is important to consult your doctor if you develop a rash. Your doctor will advise you how to deal with your symptoms or whether PREZISTA must be stopped.
Tell your doctor if you experience the following side effects:
- inflammation of the pancreas
- increased blood fat or cholesterol levels
- diabetes
- symptoms of infection
Some side effects are typical for anti-HIV medicines in the same family as PREZISTA. These are:
- raised blood sugar and worsening of diabetes.
- immune reactivation syndrome. In some patients with advanced HIV infection (AIDS) and a history of opportunistic infection, signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started, including PREZISTA. In addition to the opportunistic infections, autoimmune disorders (a condition that occurs when the immune system attacks health body tissue) may also occur after you start taking medicines for treatment of your HIV infection. Autoimmune disorders may occur many months after the start of treatment.
- increased bleeding in patients with haemophilia.
- muscle pain, tenderness or weakness. On rare occasions, these muscle disorders have been serious.
If you experience any of these side effects and they worry you, or if you notice any side effects not listed in this leaflet, please tell your doctor.
Tell your doctor if you notice signs or symptoms of infections, such as a fever or rashes. Some people with HIV who have had infections in the past may experience a return of symptoms soon after taking anti-HIV medicines.
If you think you are having an allergic reaction to PREZISTA, tell your doctor immediately or go to Accident and Emergency at your nearest hospital.
Symptoms usually include some or all of the following:
- rash, itching or hives on the skin
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
Other side effects not listed above may also occur in some people.
Please refer to the ritonavir or cobicistat Consumer Medicine Information for information on ritonavir or cobicistat.
Product Description
Storage
PREZISTA tablets should be kept out of reach of children, in a location where the temperature stays below 30°C.
What it looks like:
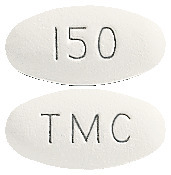
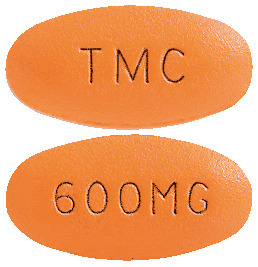
PREZISTA 600 mg film-coated tablets are orange, oval shaped tablets, with TMC on one side, 600MG on the other side. Each plastic bottle contains 60 tablets.
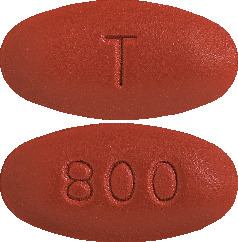
PREZISTA 800 mg film-coated tablets are dark red, oval shaped tablets, with 800 on one side and T on the other side. Each plastic bottle contains 30 tablets.
Ingredients
Active ingredient:
- darunavir 600 mg or 800 mg (as darunavir ethanolate)
Other ingredients:
- microcrystalline cellulose
- colloidal anhydrous silica
- crospovidone
- magnesium stearate
- polyvinyl alcohol
- macrogol 3350
- titanium dioxide (E171)
- purified talc
- [600 mg tablets] sunset yellow FCF aluminium lake
- [800 mg tablets] hypromellose and iron oxide red (E172).
Sponsor
JANSSEN-CILAG Pty Ltd
1-5 Khartoum Rd
Macquarie Park NSW 2113 Australia
Telephone: 1800 226 334
NZ Office: Auckland, New Zealand
Telephone: 0800 800 806

Registration numbers
600 mg tablet: AUST R 153628
800 mg tablet AUST R 199320
This leaflet was prepared in 27 March 2023.
Published by MIMS May 2023





 * Excluding laboratory abnormalities reported as ADRs.
* Excluding laboratory abnormalities reported as ADRs.
 * Excluding laboratory abnormalities reported as ADRs.
* Excluding laboratory abnormalities reported as ADRs.
 * Excluding laboratory abnormalities reported as ADRs.
* Excluding laboratory abnormalities reported as ADRs.
 * Excluding laboratory abnormalities reported as ADRs.
* Excluding laboratory abnormalities reported as ADRs.

 Rarely, events of rhabdomyolysis (associated with coadministration with HMG-CoA reductase inhibitors and Prezista) have been reported.
Rarely, events of rhabdomyolysis (associated with coadministration with HMG-CoA reductase inhibitors and Prezista) have been reported. In a pooled analysis of the POWER and DUET trials, the identified amino acid substitutions that developed on Prezista/rtv 600/100 mg b.i.d. in ≥ 20% of the isolates from patients who experienced virological failure by rebound were V32I, I54L, and L89V. Amino acid substitutions that developed in 10 to 20% of the isolates were V11I, I13V, L33F, I50V, and F53L.
In a pooled analysis of the POWER and DUET trials, the identified amino acid substitutions that developed on Prezista/rtv 600/100 mg b.i.d. in ≥ 20% of the isolates from patients who experienced virological failure by rebound were V32I, I54L, and L89V. Amino acid substitutions that developed in 10 to 20% of the isolates were V11I, I13V, L33F, I50V, and F53L. Baseline darunavir phenotype (shift in susceptibility relative to reference) was shown to be a predictive factor of virologic outcome. Response rates assessed by baseline darunavir phenotype are shown in Table 16. The data are provided to give clinicians information on the likelihood of virologic success based on pretreatment susceptibility to darunavir.
Baseline darunavir phenotype (shift in susceptibility relative to reference) was shown to be a predictive factor of virologic outcome. Response rates assessed by baseline darunavir phenotype are shown in Table 16. The data are provided to give clinicians information on the likelihood of virologic success based on pretreatment susceptibility to darunavir. In deciding on a new regimen for patients who have failed an antiretroviral regimen, careful consideration should be given to treatment history of the individual patient and the patterns of mutations associated with different agents. When available, genotypic or phenotypic testing can be performed to guide the use of darunavir.
In deciding on a new regimen for patients who have failed an antiretroviral regimen, careful consideration should be given to treatment history of the individual patient and the patterns of mutations associated with different agents. When available, genotypic or phenotypic testing can be performed to guide the use of darunavir. In the 48 week analysis, the virologic response (HIV-1 RNA < 50 copies/mL) for the Prezista/rtv arm was 83.7% and for the lopinavir/rtv arm 78.3% (see Figure 1). Statistical comparisons between the treatment arms at week 48 confirmed noninferiority of DRV/rtv versus lopinavir/rtv (p-value < 0.001) for both ITT (intent to treat) and OP (on protocol) population.
In the 48 week analysis, the virologic response (HIV-1 RNA < 50 copies/mL) for the Prezista/rtv arm was 83.7% and for the lopinavir/rtv arm 78.3% (see Figure 1). Statistical comparisons between the treatment arms at week 48 confirmed noninferiority of DRV/rtv versus lopinavir/rtv (p-value < 0.001) for both ITT (intent to treat) and OP (on protocol) population. The virological response (< 50 copies/mL) at 96 weeks by baseline viral load and baseline CD4+ cell count is presented in Table 18.
The virological response (< 50 copies/mL) at 96 weeks by baseline viral load and baseline CD4+ cell count is presented in Table 18.
 Limited data is available in patients with baseline HIV-1 RNA ≥ 100,000 copies/mL.
Limited data is available in patients with baseline HIV-1 RNA ≥ 100,000 copies/mL. In the 48 week analysis, virologic response, defined as the percentage of patients with plasma HIV-1 RNA level < 400 copies/mL, was 76.5% and 67.0% for the Prezista/rtv arm and lopinavir/rtv arm, respectively. Noninferiority in virologic response was demonstrated (p < 0.001) for both the ITT (see Figure 2) and OP population.
In the 48 week analysis, virologic response, defined as the percentage of patients with plasma HIV-1 RNA level < 400 copies/mL, was 76.5% and 67.0% for the Prezista/rtv arm and lopinavir/rtv arm, respectively. Noninferiority in virologic response was demonstrated (p < 0.001) for both the ITT (see Figure 2) and OP population.



 In women receiving darunavir/cobicistat 800/150 mg q.d. during the 2nd trimester of pregnancy, mean intra-individual values for total darunavir Cmax, AUC24h and Cmin were 49%, 56% and 92% lower, respectively, as compared with postpartum; during the 3rd trimester of pregnancy, total darunavir Cmax, AUC24h and Cmin values were 37%, 50% and 89% lower, respectively, as compared with postpartum.
In women receiving darunavir/cobicistat 800/150 mg q.d. during the 2nd trimester of pregnancy, mean intra-individual values for total darunavir Cmax, AUC24h and Cmin were 49%, 56% and 92% lower, respectively, as compared with postpartum; during the 3rd trimester of pregnancy, total darunavir Cmax, AUC24h and Cmin values were 37%, 50% and 89% lower, respectively, as compared with postpartum.
