1 Name of Medicine
Ripretinib.
2 Qualitative and Quantitative Composition
Each Qinlock tablet contains 50 mg of ripretinib.
Ripretinib is a white to off-white crystalline solid. Ripretinib is a lipophilic, weak base compound, practically insoluble in aqueous media.
Excipients with known effect.
Sugars as lactose.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
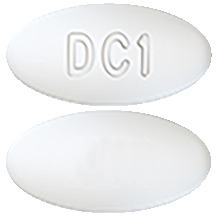
Qinlock tablets are white to off-white oval shaped tablets debossed with 'DC1' on one side.
4.1 Therapeutic Indications
Qinlock is a kinase inhibitor indicated for the treatment of adult patients with advanced gastrointestinal stromal tumours (GIST) who have received prior treatment with 3 or more kinase inhibitors, including imatinib.
4.2 Dose and Method of Administration
Dosage.
The recommended dosage of Qinlock is 150 mg (three 50 mg tablets) orally once daily with or without food until disease progression or unacceptable toxicity.
Instruct patients to swallow tablets whole.
Advise patients to take Qinlock at the same time each day.
Advise patients to take a missed dose if less than 8 hours have passed since the missed scheduled dose. If a patient misses a dose by more than 8 hours of the time it is usually taken, the patient should be instructed not to take the missed dose and simply resume the usual dosing schedule on the following day.
Advise patients not to take an additional dose if vomiting occurs after taking Qinlock and to continue with their next scheduled dose.
Dose modification guidelines.
The recommended dose reduction for adverse reactions is Qinlock 100 mg orally once daily.
Permanently discontinue Qinlock in patients who are unable to tolerate 100 mg orally once daily.
The recommended dosage modifications of Qinlock for adverse reactions are provided in Table 1.

Paediatrics.
The safety and effectiveness of Qinlock in paediatric patients have not been established.
Patients with renal impairment.
No dose adjustment is recommended for patients with mild and moderate renal impairment [creatinine clearance (CrCl 30 to 89 mL/min estimated by Cockcroft-Gault)]. The pharmacokinetics and safety of Qinlock in patients with severe renal impairment (CrCl 15 to 29 mL/min estimated by Cockcroft-Gault) have not been studied. No dosing recommendation can be made in patients with severe renal impairment.
Patients with hepatic impairment.
No dose adjustment is recommended in patients with mild (Child-Pugh A), moderate (Child-Pugh B), or severe hepatic impairment (Child-Pugh C). Data in patients with severe hepatic impairment is limited, thus close monitoring of overall safety is recommended in these patients.
Dose modifications for CYP3A inducers.
Avoid concomitant strong or moderate CYP3A inducers during Qinlock treatment.
If co-administration of a moderate CYP3A inducer can't be avoided, increase the Qinlock dosing frequency to twice daily during the co-administration period (e.g. from the recommended dose of 150 mg once daily to 150 mg twice daily). Monitor for clinical response and tolerability. If the concomitant moderate CYP3A inducer is discontinued, reduce the dose back to once daily, 14 days after the discontinuation of the moderate CYP3A inducer [see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions].
If co-administration of a strong CYP3A inducer absolutely can't be avoided, the above approach could be considered, but must be balanced against a risk of reduced efficacy due to reduced exposure [see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions]. Warn patients of this risk, and monitor for clinical response and tolerability. For patients who need to take Qinlock twice daily: advise patients to take a missed dose if less than 4 hours have passed since they were meant to take it. Otherwise, leave it as a missed dose and just take the next dose as scheduled.4.3 Contraindications
Use of Qinlock is contraindicated in patients with hypersensitivity to ripretinib or to any other component of Qinlock tablets.
4.4 Special Warnings and Precautions for Use
Palmar-plantar erythrodysaesthesia syndrome.
In the double-blind period of a randomised, placebo-controlled phase 3 trial (INVICTUS), Grade 1-2 palmar-plantar erythrodysaesthesia syndrome (PPES) occurred in 21% of the 85 patients who received Qinlock [see Section 4.8 Adverse Effects (Undesirable Effects)]. PPES led to dose discontinuation in 1.2% of patients, dose interruption in 2.4% of patients, and dose reduction in 1.2% of patients.
Based on severity, withhold Qinlock and then resume at same or reduced dose [see Section 4.2 Dose and Method of Administration].
New primary cutaneous malignancies.
In INVICTUS, cutaneous squamous cell carcinoma (cuSCC) occurred in 4.7% of the 85 patients who received Qinlock, with a median time to event of 4.6 months (range: 3.8 to 6 months). In the pooled safety population, cuSCC and keratoacanthoma occurred in 7% and 1.9% of 351 patients, respectively.
In INVICTUS, melanoma occurred in 2.4% of the 85 patients who received Qinlock. In the pooled safety population, melanoma occurred in 0.9% of 351 patients.
Perform dermatologic evaluations when initiating Qinlock and routinely during treatment. Manage suspicious skin lesions with excision and dermatopathologic evaluation. Continue Qinlock at the same dose.
Hypertension.
In INVICTUS, Grade 1-3 hypertension occurred in 14% of the 85 patients who received Qinlock, including Grade 3 hypertension in 7% [see Section 4.8 Adverse Effects (Undesirable Effects)].
Do not initiate Qinlock in patients with uncontrolled hypertension. Adequately control blood pressure prior to initiating Qinlock. Monitor blood pressure as clinically indicated during treatment with Qinlock and initiate or adjust antihypertensive therapy as appropriate.
Based on severity, withhold Qinlock and then resume at same or reduced dose or permanently discontinue [see Section 4.2 Dose and Method of Administration].
Cardiac failure.
In INVICTUS, cardiac failure occurred in 1.2% of the 85 patients who received Qinlock. In the pooled safety population, cardiac failure (cardiac failure, cardiac failure acute, acute left ventricular failure, and diastolic dysfunction) occurred in 1.7% of 351 patients, including Grade 3 adverse reactions in 1.1%.
In INVICTUS, Grade 3 decreased ejection fraction occurred in 2.6% of the 77 patients who received Qinlock and who had a baseline and at least one post-baseline echocardiogram. In the pooled safety population, Grade 3 decreased ejection fraction occurred in 3.4% of the 263 patients who received Qinlock and who had a baseline and at least one post-baseline echocardiogram.
In INVICTUS, cardiac dysfunction led to dose discontinuation in 1.2% of the 85 patients who received Qinlock. The safety of Qinlock has not been assessed in patients with a baseline ejection fraction below 50%.
Assess ejection fraction by echocardiogram or MUGA scan prior to initiating Qinlock and during treatment, as clinically indicated. Permanently discontinue Qinlock for Grade 3 or 4 left ventricular systolic dysfunction [see Section 4.2 Dose and Method of Administration].
See Section 4.6 Fertility, Pregnancy and Lactation for potential embryo-fetal toxicity.
Risk of impaired wound healing.
Impaired wound healing complications can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, Qinlock has the potential to adversely affect wound healing.
Withhold Qinlock for at least 1 week prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of Qinlock after resolution of wound healing complications has not been established.
Phototoxicity.
Due to the risk of phototoxicity associated with the use of Qinlock, it is recommended to advise patients to avoid or minimise exposure to direct sunlight, sunlamps, and other sources of ultraviolet radiation during treatment with Qinlock and for at least one week after discontinuation of treatment. Patients should be instructed to use measures such as protective clothing (long sleeves and hat) and sunscreen with high sun protection factor (SPF).
Use in the elderly.
Of the 85 patients in INVICTUS who received Qinlock 150 mg orally once daily, 24% were between 65 to 74 years of age and 9% were 75 years of age or older. Clinical studies of Qinlock did not include sufficient numbers of patients aged 65 and older to determine whether they respond differently from younger patients.
Paediatric use.
The safety and effectiveness of Qinlock in paediatric patients have not been established.
Effects on laboratory tests.
See Section 4.8 Adverse Effects (Undesirable Effects).4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of other medications on Qinlock.
Since both ripretinib and its active metabolite are mainly cleared by CYP3A, plasma concentrations of ripretinib and the active metabolite DP-5439 are increased by CYP3A inhibitors and decreased by CYP3A inducers.
Effect of strong CYP3A inhibitors.
Coadministration of Qinlock with itraconazole (a strong CYP3A inhibitor) increased ripretinib Cmax by 36% and AUC0-∞ by 99%. Use strong CYP3A inhibitors with caution and monitor for increased toxicity.
Effect of strong CYP3A inducers.
Coadministration of Qinlock with rifampin (a strong CYP3A inducer) decreased ripretinib Cmax by 18% and AUC0-∞ by 61% and also decreased DP-5439 AUC0-∞ by 57% with increased Cmax by 37% [see Section 4.2 Dose and Method of Administration].
In the presence of a strong CYP3A inducer, a doubled ripretinib dose (twice daily rather than once daily), is predicted to result in a 40% reduction in combined AUC of ripretinib and active metabolite DP-5439, compared to the usual recommended once daily dose with no inducer present.
Avoid concomitant strong CYP3A inducers during Qinlock treatment [see Section 4.2 Dose and Method of Administration].
Effect of moderate CYP3A inducers.
Coadministration of Qinlock with efavirenz (a moderate CYP3A inducer) was predicted to decrease ripretinib Cmax by 24% and decrease AUC0-∞ by 56% [see Section 4.2 Dose and Method of Administration].
In the presence of a moderate CYP3A inducer, a doubled ripretinib dose (twice daily rather than once daily), is predicted to result in a 17% reduction in combined AUC of ripretinib and active metabolite DP-5439, compared to the usual recommended once daily dose with no inducer present.
Avoid concomitant moderate CYP3A inducers during Qinlock treatment [see Section 4.2 Dose and Method of Administration].
Effect of P-gp inhibitors.
Ripretinib and its active metabolite DP-5439 are substrates of P-gp (MDR1) and BCRP, as indicated by in vitro studies. P-gp and BCRP inhibitors may increase plasma concentrations of ripretinib and its active metabolite DP-5439.
Effect of acid-reducing agents.
Coadministration of Qinlock with pantoprazole (a proton pump inhibitor) did not affect exposure to ripretinib.
Effect of Qinlock on other medications.
CYP substrates.
Ripretinib is a weak inhibitor of CYP2C8. Co-administration of Qinlock with repaglinide (a sensitive index substrate for CYP2C8) increased repaglinide AUC0-∞ by 26%. Repaglinide Cmax was unchanged.
Ripretinib and DP-5439 are not inducers of CYP1A2, CYP2B6, or CYP3A4 in vitro.
UGT1A substrates.
In vitro studies suggested that ripretinib is an inhibitor of UGT1A1, UGT1A3, UGT1A4, UGT1A7, and UGT1A8. Clinical studies with UGT1A substrates have not been conducted.
Drug transporter systems.
Ripretinib is an inhibitor of P-gp and BCRP in vitro. DP-5439 is an inhibitor of BCRP and MATE1 (Multidrug And Toxin Extrusion Protein 1) in vitro. Ripretinib may increase clinical exposures of drugs that are substrates of P-gp, BCRP, or MATE1.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Based on findings from animal studies, Qinlock may impair fertility in males of reproductive potential. Decreased testis and epididymis weights, as well as atrophy of the testes and degeneration of the seminiferous epithelium were observed in male rats at exposure levels (AUC) similar to the human exposure at 150 mg once daily.
(Category D)
There are no clinical data on the use of Qinlock in pregnant women to inform a drug-associated risk of major birth defects and miscarriage.
Based on findings from animal studies, Qinlock can cause embryo-fetal harm when administered to a pregnant woman. Qinlock should not be used during pregnancy.
Qinlock should not be used during pregnancy. In an embryo-fetal development study in which pregnant rats were administered daily doses of ripretinib during organogenesis, ripretinib was given from gestational days 6 through 18 at doses 1, 5, or 20 mg/kg/day. Dose-related malformations primarily associated with the cardiovascular and skeletal systems were observed at a dose of 20 mg/kg/day (approximately 0.4 times the human exposure at 150 mg once daily).
An increased incidence of anatomic variations, indicative of developmental toxicity, also occurred at 20 mg/kg/day. Variations included malpositioned carotid and subclavian artery origins, malpositioned subclavian artery, absent or elongated innominate artery, misshapen and nodulated ribs, bipartite, incompletely ossified, or unossified vertebral centra, small or misshapen vertebral arches, and reductions in ossified forelimb and hindlimb phalanges, hindlimb metatarsals, and caudal vertebrae were also observed at 20 mg/kg/day.
Verify the pregnancy status of females of reproductive potential prior to initiating Qinlock. Advise female patients of reproductive potential to use effective contraception during treatment and for at least 1 week after the final dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for at least 1 week after the final dose. Effects of Qinlock on contraceptive steroids have not been studied. A barrier method contraception should be added if systemic contraceptive steroids are used.
There are no data regarding the presence of ripretinib or its metabolites in human milk or their effects on the breastfed infant or on milk production. Because of the potential for adverse reactions in breastfed infants, advise women not to breastfeed during treatment with ripretinib and for at least 1 week after the final dose.4.7 Effects on Ability to Drive and Use Machines
No data available.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
In the Phase 3 double-blind, randomised (2:1), placebo-controlled trial (INVICTUS), 129 study participants with a diagnosis of advanced GIST were randomised to Qinlock (N=85) or placebo (N=44) [see Section 5.1 Pharmacodynamic Properties]. The data described in this section reflect the safety population (N=128) who had received at least one dose of Qinlock (N=85) or placebo (N=43). One study participant who was randomised to the placebo arm did not receive placebo. The safety results from the double-blind treatment period of INVICTUS are described below.
The most common adverse events (≥ 20%) observed in patients treated with Qinlock (all grades) were alopecia, fatigue, nausea, abdominal pain, constipation, myalgia, diarrhoea, decreased appetite, palmar-plantar erythrodysaesthesia syndrome (PPES), and vomiting (Table 2). The most common Grade 3 or 4 laboratory abnormalities were increased lipase and decreased phosphate (Table 3).
For additional information on palmar-plantar erythrodysaesthesia syndrome, new primary cutaneous malignancies such as squamous cell carcinoma of the skin, keratoacanthoma and melanoma, hypertension and cardiac failure (cardiac failure, cardiac failure acute, acute left ventricular failure, and diastolic dysfunction) (see Section 4.4 Special Warnings and Precautions for Use).
Tabulated list of adverse events.
Table 2 summarises the most frequently reported treatment-emergent adverse events (TEAEs) in ≥ 10% of patients with advanced GIST who received Qinlock in the double-blind treatment period of INVICTUS.
 Table 3 summarises the most frequently reported treatment-emergent laboratory abnormalities in ≥ 10% of patients with advanced GIST who received Qinlock in the double-blind treatment period of INVICTUS. Also see Table 4.
Table 3 summarises the most frequently reported treatment-emergent laboratory abnormalities in ≥ 10% of patients with advanced GIST who received Qinlock in the double-blind treatment period of INVICTUS. Also see Table 4.


Other adverse reactions.
The following additional adverse reactions have occurred in < 10% of Qinlock-treated patients in INVICTUS:
Peripheral sensory neuropathy (7.1% vs 2.3% placebo), dermatitis acneiform (5.9% vs 0% placebo), rash (4.7% vs 4.7% placebo), seborrheic keratosis (4.7% vs 0% placebo), skin papilloma (3.5% vs 0% placebo) and melanocytic naevus (2.4% vs 0% placebo).
Clinically relevant adverse reactions that occurred in < 10% of patients in the pooled safety population included cardiac ischemic events (1.1%) (including acute coronary syndrome and fatal cardiac arrest or myocardial infarction). Photosensitivity occurred in 0.6% (see Section 4.4 Special Warnings and Precautions for Use).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems and [email protected].4.9 Overdose
There is no known specific antidote for Qinlock overdose. In the event of suspected overdose, interrupt Qinlock, undertake general supportive measures, and observe until clinical stabilisation.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Ripretinib is a switch-control tyrosine kinase inhibitor with a dual mechanism of action. Ripretinib binds to both the switch pocket and the activation loop to lock the kinase in the inactive state, preventing downstream signalling and cell proliferation. This dual mechanism of action provides broad inhibition of KIT and PDGFRA kinase activity, including wild type and multiple primary and secondary mutations. Ripretinib also inhibits other kinases in vitro, such as PDGFRB, TIE2, VEGFR2, and BRAF.
Cardiac electrophysiology.
Following treatment with Qinlock 150 mg once daily, no clinically meaningful QT interval prolongation was observed.
Clinical trials.
The efficacy of Qinlock was evaluated in INVICTUS, an international, multi-centre, randomised (2:1), double-blind, placebo-controlled trial. Eligible patients had unresectable, locally advanced or metastatic gastrointestinal stromal tumour (GIST) and had received prior treatment with imatinib, sunitinib, and regorafenib. Randomisation was stratified by prior lines of therapy (3 versus ≥ 4) and Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1 or 2). Patients received Qinlock 150 mg or placebo orally once daily until disease progression or unacceptable toxicity. Tumour response assessments were performed every 28 days through for the first 4 months and then every 56 days thereafter.
The major efficacy outcome measure was progression-free survival (PFS) based on disease assessment by blinded independent central review (BICR) using modified RECIST 1.1 criteria, in which lymph nodes and bone lesions were not target lesions and a progressively growing new tumour nodule within a pre-existing tumour mass must meet specific criteria to be considered unequivocal evidence of progression. Additional efficacy outcome measures included objective response rate (ORR) by BICR and overall survival (OS). Patients randomised to receive placebo could be treated with Qinlock at the time of disease progression.
A total of 129 patients were randomised, 85 to Qinlock and 44 to placebo.
Patient characteristics of the intent-to-treat (ITT) population in INVICTUS were median age of 60 years (range: 29 to 83 years), with 39% aged ≥ 65 years; 57% were male; 75% were White; and 92% had an ECOG performance status of 0 or 1. Sixty-three percent (63%) of patients received 3 prior therapies and 37% received 4 or more prior therapies. Sixty-six percent (66%) of patients randomised to placebo switched to Qinlock after disease progression.
Efficacy results from INVICTUS are summarised in Table 5. Also see Figures 1 and 2.



5.2 Pharmacokinetic Properties
Absorption.
Ripretinib reaches peak plasma concentrations at 4 hours after a single oral dose of 150 mg ripretinib (given as three tablets each containing 50 mg). The steady state AUC0-12h observed in patients at 150 mg is 5678 nanogram.h/mL. Steady state is achieved by approximately Day 15.
Administration with a high-fat meal increased ripretinib AUC0-24 and Cmax by 30% and 22%, respectively. DP-5439 AUC0-24 and Cmax were higher by 47% and 66%, respectively.
Distribution.
Both ripretinib and its active metabolite DP-5439 bind to plasma proteins at ≥ 99%. The apparent volume of distribution (Vss/F) is approximately 307 L.
Metabolism.
Ripretinib was metabolised in vitro. CYP3A4/5 is the major metaboliser of ripretinib and its active metabolite, DP-5439, while CYP2C8 and CYP2D6 are only minor metabolisers.
Excretion.
Following oral administration of ripretinib 150 mg once daily, the mean apparent oral clearance (CL/F) of ripretinib at steady-state is 15.3 L/hr and the mean plasma elimination half-life is 14.8 hours.
In preclinical species, 14C-labeled ripretinib dosed to Sprague-Dawley rats (oral) and beagle dogs (intravenous [iv]), resulted in greater than 87% of the radioactive dose being excreted in faeces and 1.8% or less in the urine.
PK analyses obtained from urine and faeces samples in 10 healthy volunteers showed that systemic elimination of ripretinib was not primarily attributed to the kidney. Through 1 week (168 hours) after a single oral administration of 50 mg ripretinib (given alone), 0.02% of the ripretinib dose was excreted as ripretinib in urine and 34.2% of the ripretinib dose was excreted as ripretinib in faeces.
Pharmacokinetics in special patient populations.
Population pharmacokinetic analyses of demographic data indicate that no clinically meaningful differences in the pharmacokinetics of ripretinib were observed based on age (19 to 87 years), sex, race (White, Black, and Asian), body weight (39 to 138 kg), tumour type (GIST or other solid tumour), prior gastrectomy, mild to moderate renal impairment (CrCl 30 to < 90 mL/min estimated by Cockcroft-Gault) and mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin 1 to 1.5 x ULN and any AST).
The effects of severe renal impairment (CrCl 15 to 29 mL/min) on the pharmacokinetics of ripretinib have not been studied.
Patients with hepatic impairment.
In subjects with mild hepatic impairment, there were no clinically significant changes in AUC0-last and Cmax when compared to matched healthy subjects.
In subjects with moderate hepatic impairment, ripretinib AUC0-last increased 2-fold while Cmax was unchanged when compared to matched healthy subjects. The combined AUC0-last of ripretinib and DP-5439 increased 1.5-fold.
In subjects with severe hepatic impairment, ripretinib AUC0-last increased 2.6-fold and Cmax decreased by 24% when compared to matched healthy subjects. The combined AUC0-last of ripretinib and DP-5439 increased by 1.4-fold.
The observed magnitude of increase in ripretinib and the combination of ripretinib and DP-5439 exposures in subjects with hepatic impairment is unlikely to be clinically relevant based on the known safety profile of ripretinib.
The unbound fraction for both ripretinib and DP-5439 showed high variability and no clear trend was apparent between protein binding and the degrees of hepatic impairment.
5.3 Preclinical Safety Data
Genotoxicity.
Ripretinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay or clastogenic in an in vivo rat bone marrow micronucleus assay. Ripretinib was weakly positive in an in vitro clastogenicity assay in human lymphocytes without metabolic activation. Ripretinib's active metabolite (DP-5439) was not mutagenic in an in vitro bacterial reverse mutation test or clastogenic in an in vitro chromosomal aberration assay in isolated human lymphocytes. Ripretinib is not expected to pose a genotoxic risk.
Carcinogenicity.
Carcinogenicity studies have not been conducted with ripretinib.
Phototoxicity.
Ripretinib indicates a potential for photoirritation/phototoxicity based on absorption in the UV visible range (above 290 nanometer). In vitro phototoxicity assessment in 3T3 mouse fibroblast cells suggest that ripretinib exhibits a potential for phototoxicity at clinically relevant concentrations following exposure to UVA and UVB radiation.6 Pharmaceutical Particulars
6.1 List of Excipients
Each Qinlock tablet contains the following inactive ingredients: crospovidone; hypromellose acetate succinate; lactose monohydrate; magnesium stearate; microcrystalline cellulose; silicon dioxide.
6.2 Incompatibilities
No incompatibilities have been identified.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C. Protect from light.
6.5 Nature and Contents of Container
Qinlock 50 mg tablets are packaged with silica gel desiccant into white high-density polyethylene (HDPE) bottles. The bottles are closed with polypropylene child resistant closures with a polyethylene-faced induction heat seal liner. Each HDPE bottle contains 90 tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
The chemical structure of ripretinib is shown below:
 Chemical name: 1-(4-bromo-5-[1-ethyl-7-(methylamino)-2-oxo-1,2-dihydro-1,6-naphthyridin-3-yl]-2-fluorophenyl)-3-phenylurea.
Chemical name: 1-(4-bromo-5-[1-ethyl-7-(methylamino)-2-oxo-1,2-dihydro-1,6-naphthyridin-3-yl]-2-fluorophenyl)-3-phenylurea.
Molecular formula: C24H21BrFN5O2.
Molecular weight: 510.36 g/mol.
CAS number.
1442472-39-0.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes