What is in this leaflet
This leaflet answers some common questions about REMSIMA® for subcutaneous (under the skin) injection. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using REMSIMA® against the benefits it is expected to have for you.
If you have any concerns about being given this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
What REMSIMA® is used for
REMSIMA® contains the active ingredient, infliximab. Infliximab is a monoclonal antibody that is produced from human and mouse proteins by recombinant technology. Monoclonal antibodies are proteins that recognise and bind to certain special proteins in the body.
Infliximab acts by binding to a special protein in the body called tumour necrosis factor alpha (TNFα). In people with diseases such as Crohn's disease, ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis and psoriasis, the body produces too much TNFα, which can cause the body's immune system to attack normal healthy parts of the body.
REMSIMA® can block the damage caused by too much TNFα.
Rheumatoid arthritis
Rheumatoid arthritis is an inflammatory disease of the joints. REMSIMA® is used to reduce the signs and symptoms of rheumatoid arthritis and to prevent damage to the joints. You will also be given a disease-modifying medicine called methotrexate.
Ankylosing Spondylitis
Ankylosing spondylitis is an inflammatory disease of the spine. REMSIMA® can reduce the signs and symptoms of ankylosing spondylitis, thereby improving physical function.
Psoriatic arthritis
Psoriatic arthritis is an inflammatory disease of the joints in which psoriasis usually occurs in association with arthritis. Often the fingers and toes are affected, although it may occur in other parts of the body. REMSIMA® is used to reduce the signs and symptoms of psoriatic arthritis and improve the physical function in adults who have not responded well enough to previous treatments with other disease-modifying anti-rheumatic drugs (DMARDS).
REMSIMA® may be given alone or in combination with methotrexate.
Psoriasis
Psoriasis is an inflammatory disease of the skin. REMSIMA® is used to treat patients with moderate to severe psoriasis who have not responded well enough to treatments such as phototherapy or conventional systemic treatments, or when these treatments are not appropriate.
Crohn's disease
Crohn's disease is a chronic inflammatory disease of the bowel. It may also affect any part of the gut. REMSIMA® is used to treat moderate to severe Crohn's disease in adult patients who have not responded well enough to other treatments.
REMSIMA® can also reduce the number of abnormal openings from the bowel through the skin (called draining enterocutaneous fistula), a common complication of Crohn's disease.
Ulcerative Colitis
Ulcerative colitis is an inflammatory disease of the bowel. REMSIMA® is used to treat the signs and symptoms of ulcerative colitis in adult patients who have not responded well enough to other treatments.
Do not give this medicine to children and adolescents under 18 years of age because there are no data that show that this medicine is safe and works in this age group.
Your doctor, however, may prescribe REMSIMA® for another purpose.
Ask your doctor if you have any questions about why REMSIMA® has been prescribed for you.
Before you are given REMSIMA®
When you must not be given it
Do not use REMSIMA® if you have an allergy to mouse proteins or any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction to REMSIMA® may include skin rash, hives, fatigue, wheezing, difficulty in breathing, and/or low blood pressure.
Do not use REMSIMA® if you have severe infections such as tuberculosis and infected abscesses, a repeating infection or have had repeating infections.
Do not use REMSIMA® if you are already taking another medicine for arthritis, which contains the substance called anakinra.
If you have never been given REMSIMA® ® and have congestive heart failure, you should not use it.
Before you are given it
Tell your doctor if you:
- currently have an infection, or if you are prone to infections, or if you have a history of infections
REMSIMA® may affect the normal immune response. You might get infections more easily. Some cases of serious infections, including tuberculosis (TB) and sepsis have been reported in patients treated with REMSIMA®. - have ever had or been in close contact with TB, even if you were treated for it.
- have ever had or had been in close contact with hepatitis B
Reactivation of hepatitis B have been reported in people treated with TNFα blockers. However, these reports are very rare. - have lived in or travelled to an area where fungal infections called histoplasmosis, coccidioidomycosis, or blastomycosis are common. Ask your doctor if you don't know if these infections are common in the area in which you have lived in or travelled to.
These infections are caused by fungus that can affect the lungs or other parts of your body. - have had cancer
A type of blood cancer called lymphoma has been reported in patients receiving TNF blockers. The reports are rare but are more frequent than expected for people in general. Cancers, other than lymphoma, have also been reported. - have a long history of Crohn's disease rheumatoid arthritis, ankylosing spondylitis or psoriatic arthritis, especially if you have a highly active disease and/or have been taking medicine that reduces the activity of the body's natural defences.
You may be more likely to develop infections and lymphomas than people in general, even without receiving TNF blockers such as REMSIMA®. - are pregnant or plan to become pregnant
Like most medicines, REMSIMA® is not recommended in pregnancy.
You must use adequate contraception to avoid falling pregnant. - are breast-feeding
Like most medicines, REMSIMA® is not recommended while breast-feeding. It is not known whether REMSIMA® passes into breastmilk. - have or have had a disease that affects the nervous system such as multiple sclerosis and seizures, or if you experience any numbness, weakness, tingling, or sight disturbances.
- suffer from congestive heart failure.
Steps must be taken to monitor any changes to your condition during treatment with REMSIMA®. - have ongoing blood disorders or a history of blood disorders
- are scheduled to receive any vaccines. Patients receiving REMSIMA® should not receive some types of vaccines.
Your doctor will discuss with you the benefits of using REMSIMA® against the potential risks.
Taking or being given other medicines
Tell your doctor if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may affect the way other medicines work.
Do not use REMSIMA® if you are already taking another medicine for arthritis, which contains the substance anakinra.
Tell your doctor if you are already taking another medicine for arthritis which contains the substance called abatacept.
Tell your doctor if you are receiving other treatments
- for rheumatoid arthritis; for ankylosing spondylitis; for psoriatic arthritis; for Crohn’s disease or ulcerative colitis; for psoriasis, such as phototherapy
- to prevent rejection in organ transplantation.
Tell your doctor you are taking REMSIMA® before receiving any vaccinations. Some vaccinations should not be given while you are being treated with REMSIMA®.
Your doctor or pharmacist will be able to tell you what to do when being given REMSIMA® with other medicines.
How REMSIMA® is given
- REMSIMA® is only available on prescription. REMSIMA® 120 mg solution for injection is administered by injection under the skin (subcutaneous use) only. It is important to check the product labels to ensure that the correct formulation is being given as prescribed.
- For patients with rheumatoid arthritis, your doctor or nurse will start the treatment with or without two intravenous infusions. For patients with Crohn’s disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis or psoriasis, two REMSIMA® infusion doses will be given to start your REMSIMA® treatment. The first dose of REMSIMA® will be administered under the supervision of your doctor.
- After proper training, if you feel you are well-trained and confident to inject REMSIMA® yourself, your doctor may allow you to inject subsequent doses of REMSIMA® yourself at home.
Talk to your doctor if you have any questions about giving yourself an injection.
A period of observation follows treatment.
Rheumatoid arthritis
Your doctor may start your treatment with or without two REMSIMA® intravenous infusion doses of 3 mg for every kg of body weight (given to you into a vein, usually in your arm, over a period of 2 hours). If Remsima intravenous infusion doses are given to start the treatment, they are administered 2 weeks apart via intravenous infusion.
After 4 weeks from the last intravenous infusion, you will be given REMSIMA® via injection under the skin (subcutaneous injection).
If Remsima treatment is initiated without two Remsima intravenous infusion doses, the following describes how often you will usually have this medicine after your first dose:
- 2nd dose: 1 week after your 1st dose
- 3rd dose: 2 weeks after your 1st dose
- 4th dose: 3 weeks after your 1st dose
- 5th dose: 4 weeks after your 1st dose
- Further doses: 6 weeks after your 1st dose and every 2 weeks thereafter
Ankylosing Spondylitis, Psoriatic arthritis and Psoriasis
Your doctor will start your treatment with two REMSIMA® intravenous infusion doses of 5 mg for every kg of body weight (given to you into a vein, usually in your arm, over a period of 2 hours).
They are administered 2 weeks apart via intravenous infusion.
After 4 weeks from the last intravenous infusion, you will be given REMSIMA® via injection under the skin (subcutaneous injection).
The usual recommended dose of REMSIMA® subcutaneous injection is 120 mg once every 2 weeks regardless of weight.
Crohn's disease and Ulcerative colitis
Your doctor will start your treatment with two REMSIMA® intravenous infusion doses of 5 mg for every kg of body weight (given to you into a vein, usually in your arm, over a period of 2 hours).
They are administered 2 weeks apart via intravenous infusion.
After 4 weeks from the last intravenous infusion, you will be given REMSIMA® via injection under the skin (subcutaneous injection).
The usual recommended dose of REMSIMA® subcutaneous injection is 120 mg once every 2 weeks regardless of weight.
While you are being given REMSIMA®
Things you must do
Tell your doctor, nurse or pharmacist if the medicine starts to upset you or your symptoms become worse.
Tell your doctor or dentist that you are being treated with REMSIMA® before you undergo any surgical procedures.
Tell your doctor:
- if symptoms of TB (persistent cough, weight loss, listlessness, fever), or any other infection appear. Do this immediately.
- if symptoms of hepatitis B (upset stomach, loss of appetite, vomiting, tiredness, dark yellow or brown urine, and yellow eyes or skin) appear. You must do this immediately.
- that you are taking REMSIMA® before receiving any vaccinations.
Some vaccinations should not be given while you are being treated with REMSIMA®.
You should continue to take adequate contraceptive measures to avoid pregnancy.
Your doctor will also advise you not to breastfeed.
Things to be careful of
Tell your doctor if you think you have an infection. REMSIMA® may affect the normal immune response. There is a possibility that you may be more prone to infections. You will be watched closely for signs of infection.
Tell your doctor immediately if you develop a skin rash or hives. Your doctor may discontinue REMSIMA® until the symptoms go away and then begin giving the medicine again. Symptoms will resolve with appropriate treatment.
If you suffer from congestive heart failure, tell your doctor immediately if your condition worsens.
REMSIMA® is unlikely to make you drowsy. If you are tired, do not drive a car or work with machinery.
Side effects
Tell your doctor, nurse, or pharmacist as soon as possible if you do not feel well while you are being given REMSIMA®.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Generally, patients with rheumatoid arthritis, Crohn's disease, ankylosing spondylitis, psoriatic arthritis, or psoriasis already take several medicines to treat their disease.
These medicines may themselves cause side effects.
If you get additional side effects or any new symptoms, please tell your doctor.
Ask your doctor or pharmacist to answer any questions you may have.
Do not be alarmed by the following list of possible side effects. You may not experience any of them.
During the injection of REMSIMA® the following reactions may occur:
- fever or chills
- itchiness or hives
- chest pain
- low blood pressure
- high blood pressure
- shortness of breath
Tell your doctor immediately if you notice any of the following:
- pain or tenderness in chest, muscles, joints or jaw
- swelling of the hands, feet, ankles, face, lips, mouth or throat, which may cause difficulty in swallowing or breathing
- fever
- muscle pains
- joint pains
- tiredness
- abnormal chest sounds
- rash
- itching
- symptoms that may indicate heart failure, e.g. shortness of breath, especially with exercise or lying down, or swelling of your feet
- have injection site reactions such as: redness, pain, itching, swelling, hardening of the skin, bruising, coldness, pins and needles, irritation, rash, ulcer, bleeding under the skin and scan on the skin of the injection site.
Tell your doctor or nurse as soon as possible if you notice any of the following:
- headache
- nausea or vomiting
- dizziness and light-headedness
- fatigue
- fever
- rash
- hives
- itching
- sore throat
- coughing
- hoarseness
- shortness of breath
- chest pain
- back pain
- muscle pain
- abdominal pain
- indigestion
- diarrhoea
- weight loss, muscle wasting
- problems with urination
- changes in the way your heart beats, for example, if you notice it beating faster
- flushing
- dry skin or increased sweating
- fluid retention
- new onset of psoriasis, mainly on the soles of the feet and on palms
- worsening of rheumatoid arthritis.
There have been very rare cases where people taking infliximab have developed liver problems. Signs that you could be having a problem include:
- jaundice (skin and eyes turning yellow)
- dark-brown coloured urine
- right-sided abdominal pain
- fever
- severe fatigue (tiredness).
You should contact your doctor immediately if you develop any of these symptoms.
Tell your doctor if you notice any other effects.
Most of the side effects are mild to moderate in severity. Other side effects not listed above may also occur in some patients. Some side effects may appear up to six months after the last injection.
Cancers
In clinical studies, more cancers were seen in patients who received TNF-blockers, including REMSIMA®, than patients who did not receive these treatments.
In children and adults being treated with TNF-blockers, the chances of getting lymphoma or other cancers may increase. It should be noted, however, that patients with longstanding and active rheumatoid arthritis or Crohn's disease may already have a higher risk for developing cancers even without TNF-blockers, making it difficult to estimate the risk of developing cancers in these patients. Nevertheless, the role of TNFblockers in the development of cancers cannot be excluded.
A rare type of cancer called Hepatosplenic T-cell Lymphoma (HSTCL) has been reported rarely in adolescents and young adults with Crohn's disease or ulcerative colitis who have received REMSIMA®. All of these patients were also receiving drugs known as azathioprine or 6-mercaptopurine. No cases of HSTCL have been reported in patients receiving REMSIMA® only. HSTCL often results in death. The role of TNF blockers in the development of cancers in children and adolescents remain unclear.
Talk to your doctor if you are concerned about this.
Skin cancers (melanoma, Merkel cell carcinoma, basal cell carcinoma and squamous cell carcinoma) have been reported rarely in patients treated with TNFblockers, including REMSIMA®.
Tell your doctor if you notice any new skin lesions during or after therapy or if existing lesions change appearance.
Patients with a lung disease called Chronic Obstructive Pulmonary Disease and who have a history of heavy smoking may have an increased risk for getting cancer while being treated with REMSIMA®.
After REMSIMA® has been stopped
Tell your doctor immediately if:
- you notice any of the following side effects, even if they occur several weeks after stopping treatment with REMSIMA®.
- skin rash or hives
- frequent infections
- symptoms of TB (persistent cough, weight loss, listlessness, fever), or any other infection appear.
- symptoms of hepatitis B (upset stomach, loss of appetite, vomiting, tiredness, dark yellow or brown urine, and yellow eyes or skin) appear.
These symptoms may appear several months after your last REMSIMA® treatment.
You should continue to take adequate contraceptive measures to avoid pregnancy for at least 6 months after the last injection of REMSIMA®.
Your doctor will advise you not to breastfeed for at least 6 months after your last injection of REMSIMA®.
Tell your doctor if you notice any other effects.
Storage
REMSIMA® should be stored at 2°C to 8°C (Refrigerate.) Do not use beyond the expiry date.
REMSIMA® may be stored at temperatures up to a maximum of 25°C for a single period of up to 28 days, but not exceeding the original expiry date.
The medicinal product must be discarded if not used within the 14-day period. The new expiry date must be written on the carton. Upon removal from refrigerated storage, REMSIMA® must not be returned to refrigerated storage.
REMSIMA® is for single use only.
Product description
What it looks like
REMSIMA® is a clear to opalescent, colourless to pale brown solution.
REMSIMA ® is available in the following presentations:
Single use pre-filled syringe.
Each pack contains;
- 1 pre-filled syringe with 2 alcohol pads.
- 2 pre-filled syringe with 2 alcohol pads
- 4 pre-filled syringe with 4 alcohol pads
- 6 pre-filled syringe with 6 alcohol pads
Single use pre-filled syringe with automatic needle guard.
Each pack contains;
- 1 pre-filled syringe with automatic needle guard with 2 alcohol pads.
- 2 pre-filled syringe with automatic needle guard with 2 alcohol pads.
- 4 pre-filled syringe with automatic needle guard with 4 alcohol pads.
- 6 pre-filled syringe with automatic needle guard with 6 alcohol pads.
Ingredients
Active ingredient:
infliximab (rmc) 120 mg per pre-filled syringe
Inactive ingredients:
- Acetic acid
- Sodium acetate trihydrate
- Sorbitol
- Polysorbate 80
- Water for injections
Sponsor
Celltrion Healthcare Australia Pty Ltd
Suite 13.03,
31 Market Street,
Sydney 2000, Australia
Australian Registration Number
Prefilled syringe: AUST R 326143
Prefilled syringe with automatic needle guard: AUST R 326187
Date of Preparation:
August 2021
Instructions for use
Read carefully these instructions before using the REMSIMA® syringe.
Consult your healthcare provider if you have questions about using the REMSIMA® syringe.
Important information
- Use the syringe ONLY if your healthcare provider has trained you on the right way to prepare for and to give an injection.
- Ask your healthcare provider how often you will need to give an injection.
- Rotate the injection site each time you give an injection. Each new injection site should be at least 3 cm away from the previous injection site.
- Do not use the syringe if it has been dropped or is visibly damaged. A damaged syringe may not function properly.
- Do not reuse the syringe.
- Do not shake the syringe at any time.
About the REMSIMA® syringe
Parts of the syringe (see Figure A):
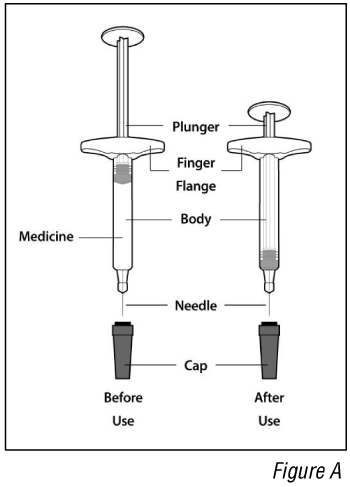
- Do not remove the cap until you are ready to inject. Once you remove the cap, do not recap the syringe.
Prepare for the injection
- Gather the supplies for the injection.
a. Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.
b. Remove the syringe from the carton stored in your refrigerator by holding the middle of the syringe body.
c. Ensure you have the following supplies:
- Syringe
- Alcohol swab
- Cotton ball or gauze*
- Adhesive bandage*
- Sharps disposal container*
*Items not included in the carton.
- Inspect the syringe.
Do not use the syringe if:
- It is cracked or damaged.
- The expiration date has passed.
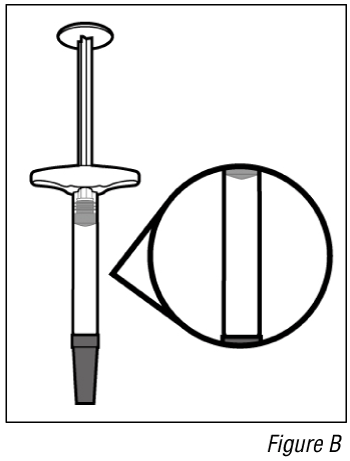
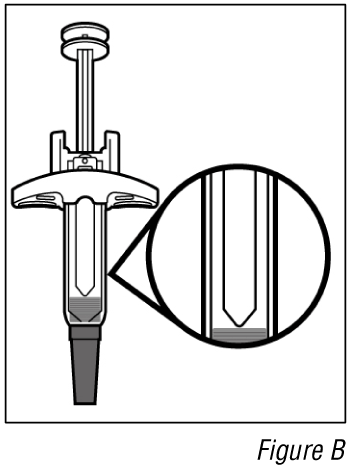
- Inspect the medicine (see Figure B).
Do not use the syringe if the liquid is different to clear colourless or pale brown or contains particles in it.
Note: You may see air bubbles in the liquid. This is normal.
- Wait 30 minutes.
a. Leave the syringe at room temperature for 30 minutes to allow it to naturally warm up.
Do not warm the syringe using heat sources such as hot water or a microwave.
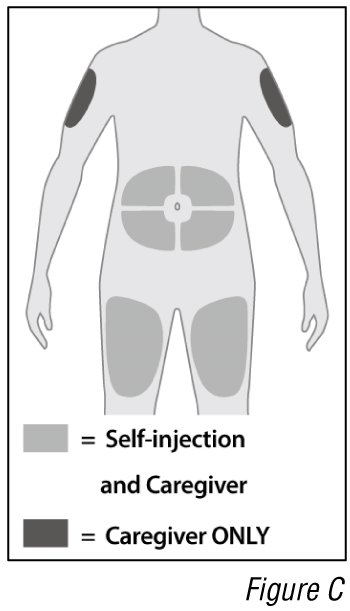
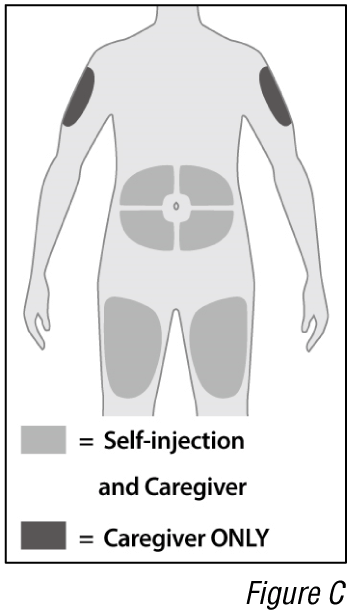
- Choose an injection site (see Figure C).
a. Select an injection site. You may inject into:
- The front of the thighs.
- The abdomen except for the 5 cm around the belly button (navel).
- The outer area of the upper arms (caregiver ONLY).
Do not inject into skin that is within 5 cm of your belly button (navel), or is tender, damaged, bruised, or scarred.
Note: Rotate the injection site each time you give an injection. Each new injection site should be at least 3 cm away from the previous injection site.
- Wash your hands.
a. Wash your hands with soap and water and dry them thoroughly.
- Clean the injection site.
a. Clean the injection site with an alcohol swab.
b. Let the skin dry before injecting.
Do not blow on or touch the injection site again before giving the injection.
Give the injection
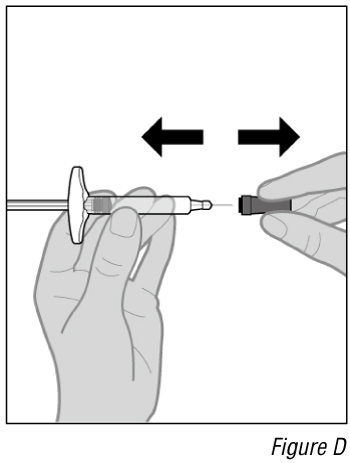
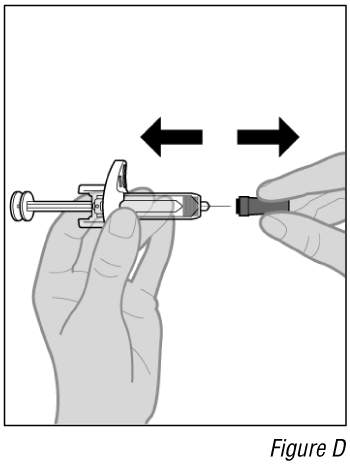
- Remove the cap (see Figure D).
a. Pull the cap straight off and set it aside.
Do not touch the needle. Doing so may result in a needle stick injury.
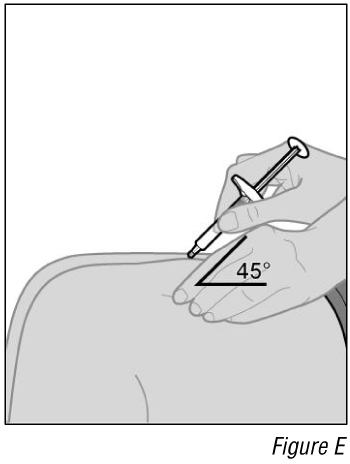
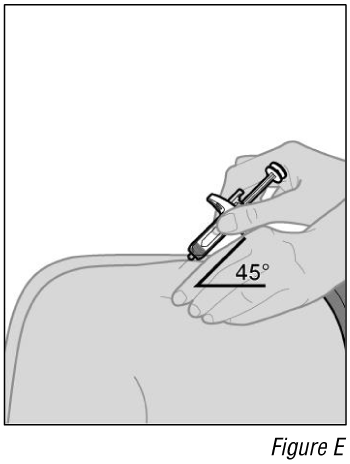
- Insert the syringe into the injection site (see Figure E).
a. Hold the syringe by its body in one hand between your thumb and index finger.
b. Using your other hand, gently pinch a fold of skin you cleaned.
c. With a quick and “dart-like” motion, insert the needle completely into the fold of the skin at a 45-degree angle.
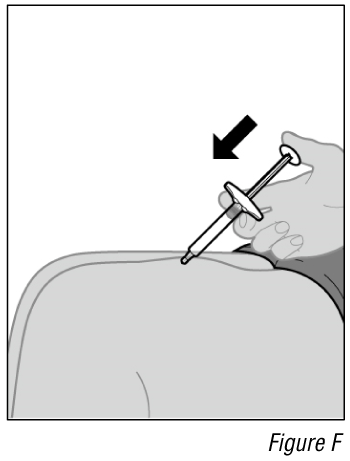
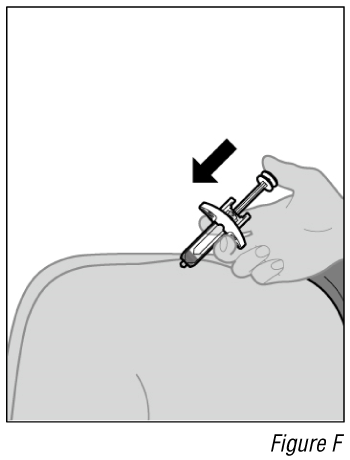
- Give the injection (see Figure F).
a. After the needle is inserted, let go of the pinched skin.
b. Push the plunger down slowly and as far as it will go until the syringe is empty.
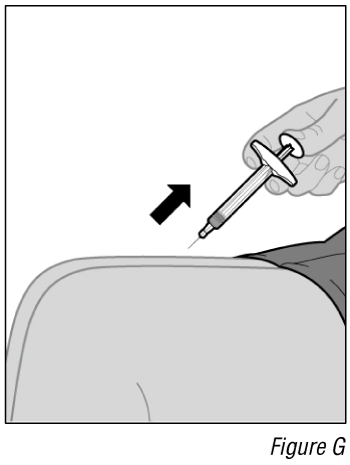
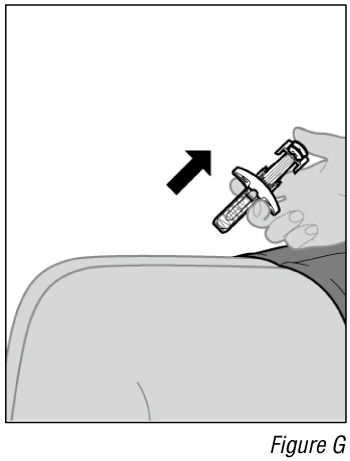
- Remove the needle from the injection site (see Figure G).
a. Remove the needle from the skin at the same angle it was inserted.
b. Gently press a cotton ball or gauze over the injection site and hold for 10 seconds.
c. Apply an adhesive bandage, if necessary.
Do not rub the injection site.
After the injection
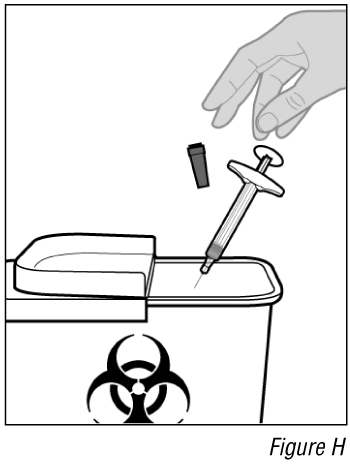
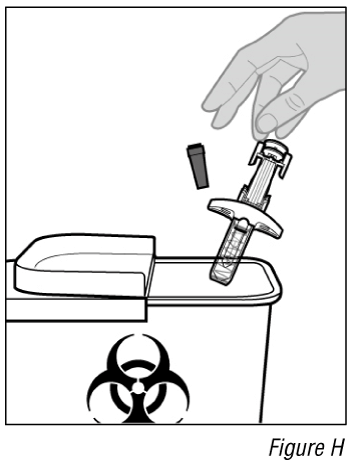
- Dispose of the syringe (see Figure H).
a. Put the used syringe in an approved sharps disposal container immediately after use.
b. If you do not have an approved sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic;
- able to close with a tight-fitting, puncture-resistant lid, without sharps being able to come out;
- upright and stable during use;
- leak-resistant; and
- properly labelled to warn of hazardous waste inside the container.
c. When your sharps disposal container is almost full, it should be disposed of in accordance with local requirements.
Do not recap the syringe.
Note: Keep the syringe and sharps disposal container out of the sight and reach of children.
Instructions for use
Read carefully these instructions before using the REMSIMA® syringe. Consult your healthcare provider if you have questions about using the REMSIMA® syringe.
Important information
- Use the syringe ONLY if your healthcare provider has trained you on the right way to prepare for and to give an injection.
- Ask your healthcare provider how often you will need to give an injection.
- Rotate the injection site each time you give an injection. Each new injection site should be at least 3 cm away from the previous injection site.
- Do not use the syringe if it has been dropped or is visibly damaged. A damaged syringe may not function properly.
- Do not reuse the syringe.
- Do not shake the syringe at any time.
About the REMSIMA® syringe
Parts of the syringe (see Figure A):
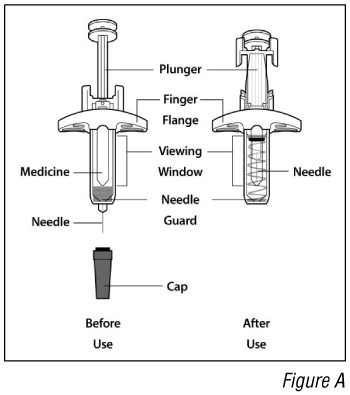
- Do not remove the cap until you are ready to inject. Once you remove the cap, do not recap the syringe.
Prepare for the injection
- Gather the supplies for the injection.
a. Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.
b. Remove the syringe from the carton stored in your refrigerator by holding the middle of the syringe body.
c. Ensure you have the following supplies:
- Syringe
- Alcohol swab
- Cotton ball or gauze*
- Adhesive bandage*
- Sharps disposal container*
*Items not included in the carton.
- Inspect the syringe.
Do not use the syringe if:
- It is cracked or damaged.
- The expiration date has passed.
- Inspect the medicine (see Figure B).
Do not use the syringe if the liquid is different to clear colourless or pale brown or contains particles in it.
Note: You may see air bubbles in the liquid. This is normal.
- Wait 30 minutes.
a. Leave the syringe at room temperature for 30 minutes to allow it to naturally warm up.
Do not warm the syringe using heat sources such as hot water or a microwave.
- Choose an injection site (see Figure C).
a. Select an injection site. You may inject into:
- The front of the thighs.
- The abdomen except for the 5 cm around the belly button (navel).
- The outer area of the upper arms (caregiver ONLY).
Do not inject into skin that is within 5 cm of your belly button (navel), or is tender, damaged, bruised, or scarred.
Note: Rotate the injection site each time you give an injection. Each new injection site should be at least 3 cm away from the previous injection site.
- Wash your hands.
a. Wash your hands with soap and water and dry them thoroughly.
- Clean the injection site.
a. Clean the injection site with an alcohol swab.
b. Let the skin dry before injecting.
Do not blow on or touch the injection site again before giving the injection.
Give the injection
- Remove the cap (see Figure D).
a. Pull the cap straight off and set it aside.
Do not touch the needle. Doing so may result in a needle stick injury.
- Insert the syringe into the injection site (see Figure E).
a. Hold the syringe by its body in one hand between your thumb and index finger.
b. Using your other hand, gently pinch a fold of skin you cleaned.
c. With a quick and “dart-like” motion, insert the needle completely into the fold of the skin at a 45-degree angle.
- Give the injection (see Figure F).
a. After the needle is inserted, let go of the pinched skin.
b. Push the plunger down slowly and as far as it will go until the syringe is empty.
- Remove the syringe from the injection site (see Figure G).
a. After the syringe is empty, slowly lift your thumb from the plunger until needle is completely covered by the automatic needle guard.
b.Gently press a cotton ball or gauze over the injection site and hold for 10 seconds.
c. Apply an adhesive bandage, if necessary.
Do not rub the injection site.
After the injection
- Dispose of the syringe (see Figure H).
a. Put the used syringe in an approved sharps disposal container immediately after use.
b. If you do not have an approved sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic;
- able to close with a tight-fitting, puncture-resistant lid, without sharps being able to come out;
- upright and stable during use;
- leak-resistant; and
- properly labelled to warn of hazardous waste inside the container.
c. When your sharps disposal container is almost full, it should be disposed of in accordance with local requirements.
Do not recap the syringe.
Note: Keep the syringe and sharps disposal container out of the sight and reach of children.
Published by MIMS November 2022

 The incidence rates of adverse events of infections in controlled and extension studies with Remsima are summarised in Table 3.
The incidence rates of adverse events of infections in controlled and extension studies with Remsima are summarised in Table 3. In extension studies following a single-way transition from Remicade to Remsima in patients with rheumatoid arthritis and ankylosing spondylitis who were treated with Remsima and Remicade for 54 weeks in controlled studies CT-P13 1.1 and CT-P13 3.1, the safety and immunogenicity profile remained consistent and similar between patients who were switched on Remsima and those who were maintained on Remsima.
In extension studies following a single-way transition from Remicade to Remsima in patients with rheumatoid arthritis and ankylosing spondylitis who were treated with Remsima and Remicade for 54 weeks in controlled studies CT-P13 1.1 and CT-P13 3.1, the safety and immunogenicity profile remained consistent and similar between patients who were switched on Remsima and those who were maintained on Remsima. Vomiting and elevated hepatic transaminases were also reported.
Vomiting and elevated hepatic transaminases were also reported. In postmarketing spontaneous reporting, Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, paediatric malignancy, leukaemia and vasculitis have been reported rarely or very rarely. During or within 2 hours of infliximab infusion transient visual loss, myocardial ischaemia/myocardial infarction and arrhythmia have occurred. Fatal and non-fatal cases of cerebrovascular accidents and myocardial ischaemia/infarction have been reported within 24 hours of infusion. There have also been reports of arrhythmia occurring within 24 hours of infusion. In addition, interstitial lung disease (including pulmonary fibrosis/interstitial pneumonitis) has been rarely observed, which in some cases may be very rarely rapidly aggressive.
In postmarketing spontaneous reporting, Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, paediatric malignancy, leukaemia and vasculitis have been reported rarely or very rarely. During or within 2 hours of infliximab infusion transient visual loss, myocardial ischaemia/myocardial infarction and arrhythmia have occurred. Fatal and non-fatal cases of cerebrovascular accidents and myocardial ischaemia/infarction have been reported within 24 hours of infusion. There have also been reports of arrhythmia occurring within 24 hours of infusion. In addition, interstitial lung disease (including pulmonary fibrosis/interstitial pneumonitis) has been rarely observed, which in some cases may be very rarely rapidly aggressive. Table 7 and Table 8 shows the proportion of patients with elevated ALT and AST in Remsima Clinical Trials.
Table 7 and Table 8 shows the proportion of patients with elevated ALT and AST in Remsima Clinical Trials.
 The difference in rates of ALT and AST elevations between Remsima and Remicade treatment groups are seen in Table 7 and Table 8.
The difference in rates of ALT and AST elevations between Remsima and Remicade treatment groups are seen in Table 7 and Table 8.
 ASPIRE trial evaluated responses at 54 weeks in 1004 methotrexate naive patients with early (≤ 3 years disease duration) active rheumatoid arthritis. Patients randomised had a median age of 51 years with a median disease duration of 0.6 years, and median swollen and tender joint count of 19 and 31, respectively. All patients received methotrexate (optimised to 20 mg/wk by week 8) and either placebo, 3 mg/kg or 6 mg/kg infliximab at weeks 0, 2, and 6 and every 8 weeks thereafter. Results from week 54 are shown in Table 11.
ASPIRE trial evaluated responses at 54 weeks in 1004 methotrexate naive patients with early (≤ 3 years disease duration) active rheumatoid arthritis. Patients randomised had a median age of 51 years with a median disease duration of 0.6 years, and median swollen and tender joint count of 19 and 31, respectively. All patients received methotrexate (optimised to 20 mg/wk by week 8) and either placebo, 3 mg/kg or 6 mg/kg infliximab at weeks 0, 2, and 6 and every 8 weeks thereafter. Results from week 54 are shown in Table 11. Data to support infliximab dose adjustment in rheumatoid arthritis comes from both ATTRACT and ASPIRE, as well as from the START study. START was a randomised, multicentre, double-blind, 3-arm, parallel-group safety study. In one of the arms the secondary objective was to assess the safety and efficacy of dose escalation above 3 mg/kg of infliximab in 1.5 mg/kg increments to a maximum of 9 mg/kg, given every 8 weeks in subjects with an inadequate response to 3 mg/kg at week 22 or if a flare occurred later. Results are shown in Table 12.
Data to support infliximab dose adjustment in rheumatoid arthritis comes from both ATTRACT and ASPIRE, as well as from the START study. START was a randomised, multicentre, double-blind, 3-arm, parallel-group safety study. In one of the arms the secondary objective was to assess the safety and efficacy of dose escalation above 3 mg/kg of infliximab in 1.5 mg/kg increments to a maximum of 9 mg/kg, given every 8 weeks in subjects with an inadequate response to 3 mg/kg at week 22 or if a flare occurred later. Results are shown in Table 12.

 There are no clinical trials with Remsima 120 mg given subcutaneously without intravenous loading doses of infliximab in patients with rheumatoid arthritis. However, population pharmacokinetic and pharmacokinetic/pharmacodynamic modelling and simulation predicted comparable infliximab exposure (AUC over 8 weeks) and efficacy (DAS28 and ACR20 response) from week 6 onward in rheumatoid arthritis patients treated with Remsima 120 mg given without intravenous loading doses of infliximab when compared with Remsima 3 mg/kg given intravenously at Weeks 0, 2 and 6, and then every 8 weeks.
There are no clinical trials with Remsima 120 mg given subcutaneously without intravenous loading doses of infliximab in patients with rheumatoid arthritis. However, population pharmacokinetic and pharmacokinetic/pharmacodynamic modelling and simulation predicted comparable infliximab exposure (AUC over 8 weeks) and efficacy (DAS28 and ACR20 response) from week 6 onward in rheumatoid arthritis patients treated with Remsima 120 mg given without intravenous loading doses of infliximab when compared with Remsima 3 mg/kg given intravenously at Weeks 0, 2 and 6, and then every 8 weeks. Furthermore, it showed that among ACR20 responders, infliximab 3 mg/kg plus methotrexate improved anaemia significantly (p = 0.034) better than methotrexate alone. Improvement in haemoglobin significantly correlated with improvement in physical function and quality of life at week 22.
Furthermore, it showed that among ACR20 responders, infliximab 3 mg/kg plus methotrexate improved anaemia significantly (p = 0.034) better than methotrexate alone. Improvement in haemoglobin significantly correlated with improvement in physical function and quality of life at week 22. In IMPACT and IMPACT 2, clinical responses were observed as early as week 2 and were maintained through week 98 and week 54, respectively. The responses were similar regardless of concomitant use of methotrexate.
In IMPACT and IMPACT 2, clinical responses were observed as early as week 2 and were maintained through week 98 and week 54, respectively. The responses were similar regardless of concomitant use of methotrexate. Infliximab-treated patients also demonstrated significant improvement in physical function as assessed by HAQ. Significant improvements in health-related quality of life were also demonstrated as measured by the physical and mental component summary scores of the SF-36 in IMPACT 2.
Infliximab-treated patients also demonstrated significant improvement in physical function as assessed by HAQ. Significant improvements in health-related quality of life were also demonstrated as measured by the physical and mental component summary scores of the SF-36 in IMPACT 2. At week 10, 82.9% of infliximab patients achieved a PGA score of minimal or cleared compared to 3.9% of placebo patients (p < 0.001). PGA scores at weeks 6, 10, 24 and 50 are presented in Table 19.
At week 10, 82.9% of infliximab patients achieved a PGA score of minimal or cleared compared to 3.9% of placebo patients (p < 0.001). PGA scores at weeks 6, 10, 24 and 50 are presented in Table 19. The median baseline value for the DLQI was 12.5. The mean baseline values were 45.6 for the SF-36 physical component and 45.7 for the mental component. Quality of life improved significantly compared to placebo at weeks 10 and 24 when evaluated by both DLQI and SF-36.
The median baseline value for the DLQI was 12.5. The mean baseline values were 45.6 for the SF-36 physical component and 45.7 for the mental component. Quality of life improved significantly compared to placebo at weeks 10 and 24 when evaluated by both DLQI and SF-36. In the multidose trial, 573 patients, with a score of at least CDAI 220, received 5 mg/kg at week 0. After assessment of response, patients were randomly assigned to one of three treatment groups; placebo at weeks 2 and 6 and then every 8 weeks until week 46; 5 mg/kg at weeks 2 and 6, and then every 8 weeks; and the 10 mg/kg maintenance group, which received 5 mg/kg at weeks 2 and 6, and then 10 mg/kg every 8 weeks. The prespecified co-primary endpoints were the proportion of patients who responded at week 2 and were in remission (CDAI < 150) at week 30 and the time to loss of response in patients who responded. Analyses of the endpoints were on the intent to treat patient population.
In the multidose trial, 573 patients, with a score of at least CDAI 220, received 5 mg/kg at week 0. After assessment of response, patients were randomly assigned to one of three treatment groups; placebo at weeks 2 and 6 and then every 8 weeks until week 46; 5 mg/kg at weeks 2 and 6, and then every 8 weeks; and the 10 mg/kg maintenance group, which received 5 mg/kg at weeks 2 and 6, and then 10 mg/kg every 8 weeks. The prespecified co-primary endpoints were the proportion of patients who responded at week 2 and were in remission (CDAI < 150) at week 30 and the time to loss of response in patients who responded. Analyses of the endpoints were on the intent to treat patient population. Significant improvement in quality of life measures were seen in both the IBDQ and SF-36 (p < 0.001) scores in infliximab-treated patients at week 30.
Significant improvement in quality of life measures were seen in both the IBDQ and SF-36 (p < 0.001) scores in infliximab-treated patients at week 30. In both studies, a significantly greater percentage of patients in the infliximab groups were in clinical response and clinical remission at week 8 when compared to placebo. Furthermore, in both ACT 1 and ACT 2, a significantly greater proportion of patients treated with 5 mg/kg or 10 mg/kg infliximab experienced clinical response and clinical remission at week 30 compared to placebo treatment. In addition, the proportion of patients in sustained response (i.e. were in clinical response at both week 8 and week 30) in the infliximab groups was at least twice as large as in the placebo group. Results from weeks 8 and 30 are shown in Table 20.
In both studies, a significantly greater percentage of patients in the infliximab groups were in clinical response and clinical remission at week 8 when compared to placebo. Furthermore, in both ACT 1 and ACT 2, a significantly greater proportion of patients treated with 5 mg/kg or 10 mg/kg infliximab experienced clinical response and clinical remission at week 30 compared to placebo treatment. In addition, the proportion of patients in sustained response (i.e. were in clinical response at both week 8 and week 30) in the infliximab groups was at least twice as large as in the placebo group. Results from weeks 8 and 30 are shown in Table 20. The following secondary PK endpoints, Cmin, Cmax and Tmax were assessed up to week 54 and these results remained comparable.
The following secondary PK endpoints, Cmin, Cmax and Tmax were assessed up to week 54 and these results remained comparable.



