What is in this leaflet
Please read this leaflet before you start using REVESTIVE.
This leaflet answers some common questions about REVESTIVE. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using REVESTIVE against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What REVESTIVE is used for
REVESTIVE is used to treat adults with Short Bowel Syndrome who need additional nutrition or fluids from intravenous feeding (parenteral support). Short Bowel Syndrome is a disorder arising from an inability to absorb food nutrients and fluid across the gastrointestinal tract (gut). It is often caused by surgical removal of all or part of the small intestine.
REVESTIVE contains the active substance teduglutide. It improves the absorption of nutrients and fluid from your remaining gut.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
Before you use REVESTIVE
When you must not use it
Do not use REVESTIVE if you have an allergy to:
- any medicine containing teduglutide
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not use REVESTIVE if you:
- have cancer in the gastrointestinal tract, liver, gallbladder, bile ducts or pancreas
- have had cancer in the gastrointestinal tract, including liver, gallbladder, bile ducts or pancreas, within the last five years.
Do not give this medicine to a person below 18 years of age. The benefits and risks in children or adolescents below 18 years of age have not been established.
Do not use this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start using this medicine, talk to your doctor.
Before you start to use it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- severely decreased liver function
- certain cardiovascular diseases (affecting the heart and/or blood vessels), such as high blood pressure or a weak heart. The symptoms include sudden weight gain, swollen ankles and/or shortness of breath
- decreased kidney function
- other severe diseases that are not well controlled.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
Medical check-ups before and during treatment
Before you start treatment with this medicine, your doctor will need to perform a colonoscopy (a procedure to see inside your colon and rectum) to check for the presence of polyps (small abnormal growths) and remove them. It is recommended that your doctor performs these examinations once a year during the first two years after starting treatment, and then at a minimum of five-year intervals. If polyps are found either before or during your treatment with REVESTIVE, your doctor will decide whether you should continue using this medicine.
REVESTIVE should not be used if a cancer is detected during your colonoscopy.
If you have not told your doctor about any of the above, tell him/her before you start using REVESTIVE.
Taking other medicines
Tell your doctor or pharmacist if you are using any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
REVESTIVE may affect how other medicines are absorbed from the gut and therefore how well they work. Your doctor may have to change your dose of other medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while using this medicine.
How to use REVESTIVE
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor, pharmacist or nurse for help.
How much to use
The recommended daily dose is 0.05 mg per kg body weight once per day, given under the skin. The dose will be prescribed as mL of solution.
Your doctor will choose the dose that is right for you depending on your body weight. Your doctor will tell you which dose to inject. If you are not sure, ask your doctor, pharmacist or nurse.
How to use it
REVESTIVE is injected under the skin (subcutaneously) once daily.
The injection can be self-administered or given by another person, for example your doctor, his/her assistant or your home nurse.
If you are injecting the medicine yourself, you must receive adequate training by your doctor or nurse. You will find detailed instructions for injections at the end of this leaflet.
How long to use it
Continue using this medicine for as long as your doctor prescribes it for you.
Do not stop using this medicine without consulting your doctor, as a sudden stop can cause changes in your fluid balance.
If you forget to use it
If you forget to inject this medicine (or cannot inject it at your usual time), use it as soon as possible on that day.
Never use more than one injection in the same day.
Do not inject a double dose to make up for the dose that you missed.
If you have trouble remembering to use your medicine, ask your pharmacist or nurse for some hints.
If you use too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (Australia: 13 11 26; New Zealand: 0800 POISON or 0800 764766) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have used too much REVESTIVE. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using REVESTIVE
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are using REVESTIVE.
Tell any other doctors, dentists, and pharmacists who treat you that you are using this medicine.
If you become pregnant while using this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked. It is recommended that your doctor performs examinations to check for the presences of polyps once a year during the first two years after starting treatment, and then at a minimum of five-year intervals. (See 'Medical check-ups before and during treatment' in 'Before you use REVESTIVE' section).
Your doctor will take special care and monitor your small bowel function and monitor for signs and symptoms indicating problems with your gallbladder, bile ducts and pancreas. Your doctor will also monitor your fluid and electrolyte balance to make sure you do not get fluid overload or dehydration.
Things you must not do
Do not use REVESTIVE to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop using your medicine or lower the dosage without checking with your doctor. If you stop using it suddenly, your body may no longer be able to absorb enough fluid and nutrients from your gut, this could lead to dehydration and imbalance of your body's electrolytes. If you need to stop this treatment for any reason, your doctor or healthcare team will advise you and monitor your condition during this period.
Things to be careful of
Be careful driving or operating machinery until you know how REVESTIVE affects you. This medicine may cause you to feel dizzy. If this happens to you, do not drive or use machines until you feel better.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are using REVESTIVE.
Like all medicines, REVESTIVE can cause some side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- cold or flu-like symptoms
- decreased appetite
- headache
- stomach pain
- bloated stomach
- polyps (small abnormal growths) in your large bowel
- flatulence
- nausea
- vomiting
- swelling of stoma (an artificial opening for waste removal)
- trouble sleeping
- cough
- urinary tract infection
- reddening, pain or swelling at the site of the injection
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor as soon as possible if you notice any of the following:
- swelling of hands, face or feet
- any infection of the sinuses, throat, airways or lungs (respiratory tract infection)
- chest pain
- tiredness, shortness of breath, with or without swelling of ankles or legs (congestive heart failure)
The above list includes serious side effects that may require medical attention. Serious side effects are rare.
Contact your doctor or go to the emergency unit immediately if any of the following occurs:
- allergic reaction which may result in a rash or more rarely to a sharp drop in blood pressure, difficulty breathing, and hives/itching (anaphylactic reaction)
- severe stomach ache, fever (inflammation of the pancreas)
- severe stomach ache, vomiting, constipation (blockage of the bowel)
- yellowing of the skin and the whites in the eyes, itching, dark urine and light-coloured stools or pain in the upper right side or middle of the stomach area (reduced flow of bile from the gallbladder and/or inflammation of the gallbladder)
Tell your doctor, pharmacist or nurse if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After using REVESTIVE
Storage
Keep your medicine in the pack until it is time to take it. If you take the medicine out of the pack it may not keep well.
Keep the medicine in a cool dry place where the temperature stays below 25°C. Do not freeze it.
After reconstitution, the solution should be used immediately. However, chemical and physical stability has been demonstrated for 3 hours at 25°C.
Do not store REVESTIVE or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop using this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What REVESTIVE looks like
REVESTIVE is a powder and solvent for solution for injection (5 mg powder in vial, 0.5 mL solvent in pre-filled syringe). The powder is white and the solvent is clear and colourless.
One vial of powder contains 5 mg of teduglutide. After reconstitution, each vial contains 5 mg teduglutide in 0.5 mL of solution, corresponding to a concentration of 10 mg/mL.
Each pack contains 28 vials with powder and 28 pre-filled syringes with solvent.
Ingredients
Active ingredient:
REVESTIVE contains teduglutide as the active ingredient.
Other ingredients:
- histidine
- mannitol
- monobasic sodium phosphate monohydrate
- dibasic sodium phosphate heptahydrate
The solvent is sterile water for injections.
Sponsor
Shire Australia Pty Limited
Shire is now part of Takeda
Level 39, 225 George Street
Sydney, NSW 2000
Australia
Telephone: 1800 012 612
This leaflet was prepared in April 2020.
Australian Registration Number:
AUST R 274911
REVESTIVE is a registered trademark of Shire-NPS Pharmaceuticals, Inc., a Takeda company.
Instructions for preparing and injecting REVESTIVE
Important information
Read the leaflet carefully before using REVESTIVE.
REVESTIVE is for injection under the skin (subcutaneous injection). Do not inject it into a vein or muscle.
After reconstitution, from a microbiological point of view, the solution should be used immediately. However, chemical and physical stability has been demonstrated for 3 hours at 25°C.
Do not use REVESTIVE if you notice that the solution is cloudy or contains particulate matter.
Dispose of all needles and syringes in a sharps disposal container.
Materials in the REVESTIVE pack:
- 28 vials with 5 mg teduglutide as a powder
- 28 pre-filled syringes with solvent
Materials needed but not included in the REVESTIVE pack:
- reconstitution needles (size 22G, length 1 ½ " (0.7 x 40 mm))
- 1 mL injection syringes (with scale intervals of 0.02 mL or smaller)
- thin injection needles for subcutaneous injection (for example, size 26G, length 5/8" (0.45 x 16 mm))
- alcohol wipes
- alcohol swabs
- a puncture-proof container for safe disposal of the used syringes and needles
NOTE: Before you start, make sure you have a clean work surface and that you have washed your hands before proceeding.
1. Prepare the pre-filled syringe
Once you have all the materials ready, you need to prepare the prefilled syringe. The following procedure shows how you do this.
1.1 Take the pre-filled syringe containing the solvent and flip off the top part of the white plastic cap on the pre-filled syringe so that it is ready for the reconstitution needle to be attached.

1.2 Attach the reconstitution needle (22G, 1½" (0.7 x 40 mm)) to the prefilled syringe by screwing it on in a clockwise direction.

2. Dissolve the powder
Now you are ready to dissolve the powder with the solvent.
2.1 Remove the green flip-off button from the powder vial, wipe the top with an alcohol wipe and allow it to dry. Do not touch the top of the vial.

2.2 Uncap the reconstitution needle on the pre-filled syringe with solvent without touching the tip of the needle.

2.3 Taking the powder vial, insert the reconstitution needle attached to the pre-filled syringe into the centre of the rubber stopper and gently push the plunger all the way down to inject all the solvent into the vial.

2.4 Leave the reconstitution needle and empty syringe in the vial. Let the vial rest for about 30 seconds.

2.5 Gently roll the vial between your palms for about 15 seconds. Then gently turn the vial upside-down once with the reconstitution needle and empty syringe still in the vial. Do not shake the vial. Shaking the vial may produce foam, which makes it difficult to extract the solution from the vial.

2.6 Let the vial rest for about 2 minutes.

2.7 Observe the vial for any undissolved powder. If any powder remains, repeat steps 2.5 and 2.6.
Do not shake the vial. If there is still some undissolved powder, discard the vial and start the preparation again from the beginning with a new vial.
NOTE: The final solution should be clear. If the solution is cloudy or contains particulate matter, do not inject it.
NOTE: Once prepared, the solution should be used immediately. It should be kept below 25°C and maximum storage time is 3 hours.
3. Prepare the injection syringe
3.1 Remove the reconstitution syringe from the reconstitution needle which is still in the vial and discard the reconstitution syringe.

3.2 Take the injection syringe and attach it to the reconstitution needle which is still in the vial.

3.3 Turn the vial upside down, slide the tip of the reconstitution needle close to the stopper and allow all the medicine to fill the syringe by pulling the plunger back gently.

NOTE: If your doctor has told you that you need two vials, prepare a second pre-filled syringe with solvent and a second powder vial as shown in the main steps 1 and 2. Withdraw the solution from the second vial into the same injection syringe by repeating step 3.
3.4 Remove the injection syringe from the reconstitution needle leaving the needle in the vial. Discard the vial and reconstitution needle together into the sharps disposal container.

3.5 Take the injection needle (26G, 5/8" (0.45 x 16 mm)), but do not remove the plastic needle cap. Attach the needle to the injection syringe containing the medicine.

3.6 Check for air bubbles. If air bubbles are present, gently tap the syringe until they rise to the top. Then gently push up the plunger to expel the air.

3.7 Your dose in mL has been calculated by your doctor. Expel any excessive volume from the syringe with the needle cap still on until your dose is reached.

4. Inject the solution
4.1 Find an area on your belly, or if you have pain or hardening of the tissue on your belly, on your thigh where it is easy for you to give the injection.
NOTE: Do not use the same area each day for each injection - rotate sites each day (use upper, lower, and left and right side of your belly) to avoid discomfort. Do not inject into the side if it has a stoma (an artificial opening for waste removal). Avoid areas that are inflamed, swollen, scarred or covered by a mole, birthmark or other lesion.


4.2 Clean the intended site of injection on your skin with an alcohol swab, using a circular motion, working outwards. Allow the area to air-dry.

4.3 Remove the plastic cap from the needle of the prepared injection syringe. Gently grasp the cleaned skin at the injection site with one hand. With the other hand, hold the syringe as you would with a pencil. Bend your wrist back and quickly insert the needle at a 45° angle.

4.4 Pull back the plunger slightly. If you see any blood in the syringe, withdraw the needle and replace the needle on the injection syringe with a clean one of the same size. You can still use the medicine that is already in the syringe. Try to inject in another place in the cleaned skin area.
4.5 Inject the medicine slowly by pushing steadily on the plunger until all the medicine is injected and the syringe is empty.
4.6 Pull the needle straight out of the skin and discard the needle and syringe together into the sharps disposal container. A small amount of bleeding may occur. If necessary, press gently on the injection site with an alcohol swab or 2x2 gauze until any bleeding has stopped.
4.7 Dispose of all needles and syringes in a sharps disposal container.
Published by MIMS June 2020
 The powder in the vial must be dissolved by adding all the solvent from the pre-filled syringe. The vial should not be shaken, but can be rolled between the palms and gently turned upside-down once.
The powder in the vial must be dissolved by adding all the solvent from the pre-filled syringe. The vial should not be shaken, but can be rolled between the palms and gently turned upside-down once.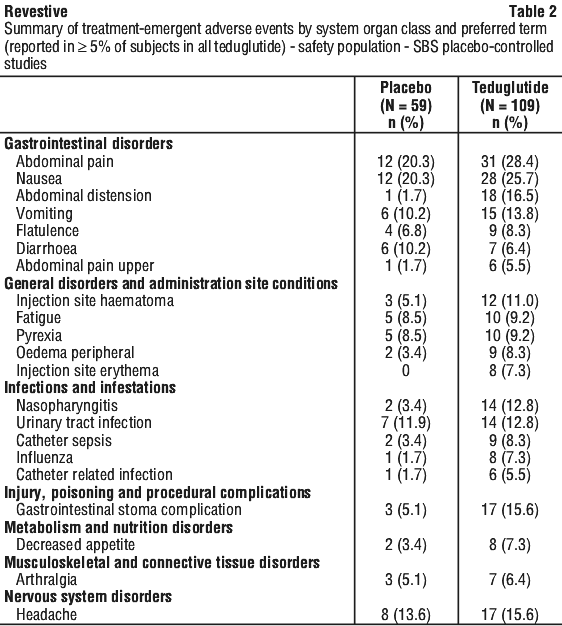 Adverse reactions are listed by MedDRA system organ class and by frequency. Frequencies are defined as very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000). Adverse reactions from post-marketing experience are indicated with #.
Adverse reactions are listed by MedDRA system organ class and by frequency. Frequencies are defined as very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000). Adverse reactions from post-marketing experience are indicated with #.
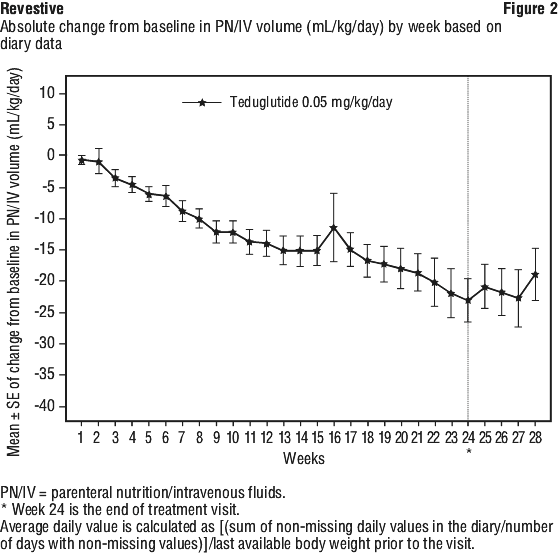
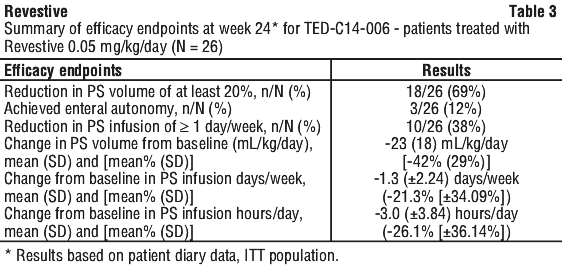 Three subjects (12%) in the 0.05 mg/kg group achieved enteral autonomy by week 24. See Figure 2.
Three subjects (12%) in the 0.05 mg/kg group achieved enteral autonomy by week 24. See Figure 2.