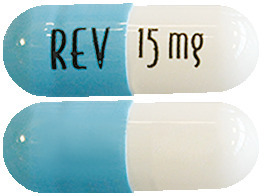


WHAT IS IN THIS LEAFLET
This leaflet answers some common questions about Revlimid. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Revlimid against the benefits this medicine is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
WHAT REVLIMID IS USED FOR
Revlimid contains an active substance called lenalidomide. Revlimid belongs to a group of medicines called immunomodulating agents that work by acting on the cells involved in the body's immune system. The immune system is part of the body's defence which helps to fight illness and infection.
Treatment of Multiple Myeloma
Multiple myeloma (MM) is a cancer of the bone marrow.
Revlimid is used to treat patients with Multiple Myeloma.
Treatment of Myelodysplastic Syndromes
Revlimid is also used to treat patients who have conditions called myelodysplastic syndromes (MDS) in whom the bone marrow does not produce enough mature blood cells. This causes a lack of healthy blood cells in the body. There are different types of MDS.
Revlimid is approved to treat a type of MDS where part of chromosome 5 is missing. This type of MDS is known as deletion 5q MDS (or 5q minus). Patients with this type of MDS often have low red blood cell counts that require treatment with blood transfusions. It is hoped that the use of Revlimid will reduce the need for blood transfusions.
Treatment of Mantle Cell Lymphoma
Revlimid is used to treat adult patients who have been diagnosed with and previously treated for Mantle Cell Lymphoma (MCL). MCL is a cancer of the lymph tissue (part of the immune system), affecting a type of white blood cell called 'B-lymphocytes'. MCL is a disease where B-cells grow in an uncontrolled way and accumulate in the lymph tissue, bone marrow or blood.
Ask your doctor if you have any questions about how Revlimid works, or why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
Revlimid will only be prescribed to you by a doctor who has experience in medicines to treat cancers of the blood.
BEFORE YOU TAKE REVLIMID
Please read the Consumer Medicine Information leaflets of any medicinal products to be taken in combination with Revlimid before starting treatment with Revlimid.
When you must not take it:
Do not take this medicine if you are pregnant, or think that you are pregnant. Revlimid may cause birth defects (deformed babies), and may affect your developing baby if you take it during pregnancy.
Do not take this medicine if you are able to become pregnant, unless you are willing to follow the required pregnancy prevention measures (outlined in Celgene's i-access® Program - see section 'Before you start to take it').
If you are not sure whether you should start taking this medicine, talk to your doctor.
Do not take Revlimid if you have an allergy to lenalidomide or any of the other ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic response may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
If you think you may be allergic to Revlimid, ask your doctor for advice.
Before you start to take it:
Follow your doctor's instructions carefully. You will have been given specific instructions by your doctor particularly on the potential effects of lenalidomide on unborn babies.
If you have not fully understood these instructions, ask your doctor again before taking Revlimid. Your doctor will have enrolled you in the i-access® Program to ensure that lenalidomide is used safely.
THE i-access® PROGRAM
Revlimid (lenalidomide) is structurally related to 'thalidomide', which is known to cause severe life-threatening human birth defects (deformed babies) and can cause death to an unborn baby if taken during pregnancy. If Revlimid is taken during pregnancy, it may cause birth defects or death to an unborn baby.
To avoid exposure to unborn babies, Revlimid has restricted availability under a Pregnancy Prevention Program (i-access®). This program is designed to ensure that this medicine is always prescribed and taken in the recommended way. Importantly, only patients who are formally enrolled in this program and agree to fully comply with all the requirements of this program can receive Revlimid.
Some of the requirements of the i-access® Program are outlined in the following sections. Your doctor will discuss all the details with you.
- FOR WOMEN TAKING REVLIMID
Before starting this treatment, your doctor will discuss your potential to become pregnant, even if you think this is unlikely e.g. if your periods have stopped.
If you are able to become pregnant:
- Your doctor will discuss the potential risk to unborn babies if Revlimid is taken during pregnancy.
- You will be required to have pregnancy tests before treatment, every 4 weeks during treatment, and 4 weeks after stopping treatment.
- You should start your Revlimid treatment as soon as you get it from the pharmacy following a negative pregnancy test.
- Use reliable means of contraception for at least 4 weeks before starting Revlimid treatment, during treatment and treatment interruption, and for at least 4 weeks after Revlimid treatment has stopped.
Your doctor will tell you what method of contraception to use.
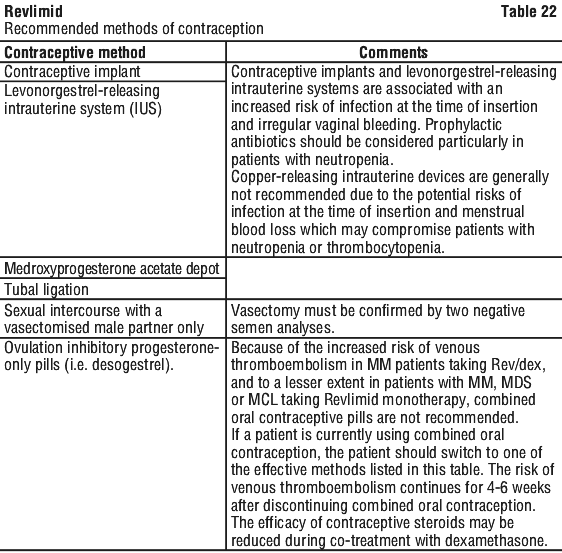
Effective methods of contraception include the following:
- Implant
- Levonorgestrel-releasing intrauterine system (IUS)
- Medroxyprogesterone acetate depot
- Tubal sterilisation
- Sexual intercourse with a vasectomised male partner only; vasectomy must be confirmed by two negative semen analyses
- Ovulation inhibitory progesterone-only pills (i.e. desogestrel).
Combined oral contraceptive pills are not recommended as they can increase the risk of blood clots blocking blood vessels in patients with MM being treated with this medicine.
You must stop taking Revlimid and inform your doctor straight away if:
- You miss or think you have missed a period, or you have unusual menstrual bleeding, or suspect you are pregnant.
- You have heterosexual intercourse without using reliable means of contraception.
Discuss with your doctor if you should breast-feed whilst taking this medicine.
It is not known if Revlimid is excreted in human milk. Therefore, you should discuss with your doctor whether to discontinue breast-feeding while you are receiving this medicine.
- FOR MEN TAKING REVLIMID
Before starting this treatment, discuss with your doctor if your partner is able to become pregnant.
If your partner is able to become pregnant, use barrier methods of contraception (e.g. condoms) even if you are vasectomised, during Revlimid treatment, during treatment interruption, and for at least 7 days after treatment has stopped.
Tell your doctor immediately if your partner becomes pregnant whilst you are taking this medicine.
Do not donate semen during treatment or during treatment interruption, or for 7 days after stopping treatment.
- FOR ALL PATIENTS TAKING REVLIMID
Discuss with your doctor if you have or have had any of the following medical conditions:
- Heart attack, blood clots, high blood pressure or high cholesterol
- Frequent bleeding or bruising
- Frequent infections
- Hepatitis B virus infection
- Peripheral neuropathy (numbness, tingling, weakness, abnormal co-ordination or pain in your hands and feet)
- Thyroid problems
- Abnormal kidney function
- Liver problems e.g. liver infections
- Allergic reactions to thalidomide or lenalidomide.
If you have not told your doctor about any of the above, tell him/her before you start taking Revlimid.
Do not donate blood during Revlimid treatment or during treatment interruption, and for at least 1 week after stopping treatment. In Australia, patients with certain cancers are permanently excluded from donating blood.
Do not take Revlimid if you have the rare hereditary problems of glucose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption. Revlimid contains lactose.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Your doctor will ask you to have regular blood tests during treatment with Revlimid. Your doctor may adjust your dose of Revlimid or stop your treatment based on the results of your blood tests and on your general condition. If you are older than 65 years, in addition to these blood tests, your doctor may also check your kidney function with other tests.
Do not give this medicine to a child or adolescent under the age of 18 years. Safety and effectiveness in children younger than 18 years have not been established.
It is important to note that a small number of patients with MM may develop additional types of cancer (regardless of their type of therapy). At this stage, it cannot be excluded that this risk may be slightly increased with Revlimid treatment. Therefore, your doctor will carefully evaluate the benefit and risk when you are prescribed this medicine.
Taking other medicines:
Tell your doctor or pharmacist if you are taking any other medicines or have recently taken any other medicines, including any medicines that you buy without a prescription from a pharmacy, supermarket or health-food shop.
Some medicines and Revlimid may interfere with each other. These include:
- medicines used to prevent pregnancy, such as oral contraceptives
- medicines used to treat symptoms of menopause e.g. hormone replacement therapy
- medicines used for heart problems e.g. digoxin
- medicines used to thin the blood e.g. warfarin.
HOW TO TAKE REVLIMID
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
How much to take:
Your doctor will tell you how much Revlimid to take and for how long you will need to take it.
For treatment of NDMM in combination with bortezomib and dexamethasone, the usual starting dose of Revlimid is 25 mg once daily. Your doctor will tell you if you are to take Revlimid for 14 continuous days of a 21-Day cycle or for 21 continuous days of a 28-Day cycle. Your doctor will also tell you the duration and the quantity of the other medicines to be taken in combination with Revlimid. After the initial treatment of about 24 weeks, you may have a stem cell transplant or your doctor may ask you to take 25 mg of Revlimid once daily for 21 days of a 28-Day cycle continuously.
For the treatment of NDMM after a stem cell transplant, the usual starting dose is 10 mg once daily continuously (28 days of a 28-Day cycle).
For the treatment of MM in combination with dexamethasone (either NDMM in patients not eligible for stem cell transplantation or MM in patients whose disease has progressed after one therapy), the usual starting dose is 25 mg once a day for 21 days of a 28-Day cycle.
For the treatment of MDS, the recommended starting dose is 10 mg once a day for 21 days of a 28-Day cycle.
For the treatment for MCL, the usual starting dose is 25 mg once a day for 21 days of a 28-Day cycle.
Your doctor will monitor your progress, and may adjust your dose of Revlimid or stop your treatment based on the results of your blood tests and on your general condition.
How to take it:
Swallow the capsules whole, preferably with water, once a day as directed by your doctor.
Do not open, break or chew the capsules.
If powder from inside the capsules leaks out and contacts the skin, wash the skin immediately and thoroughly with soap and water. If lenalidomide contacts the mucous membranes e.g. the eyes, flush thoroughly with water.
When to take it:
Take your medicine either one hour before or two hours after eating food.
How long to take it:
Continue taking Revlimid as instructed by your doctor, until your doctor tells you to stop.
Your doctor will keep a close check on you to make sure you continue to benefit from Revlimid.
If you forget to take Revlimid:
If it is less than 12 hours before your next dose, skip the dose you missed and take the next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much Revlimid (overdose):
In Australia, immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at your nearest hospital, if you think that you or anyone else may have taken too much Revlimid.
In New Zealand, immediately telephone your doctor or contact the National Poisons Centre (telephone 0800 POISON or 0800 764 766) for advice, or go to the Emergency Department at your nearest hospital, if you think that you or anyone else may have taken too much Revlimid.
Do this even if there are no signs of discomfort or poisoning.
Keep the telephone numbers for these places handy.
If you have any further questions on the use of Revlimid, ask your doctor or pharmacist.
WHILE YOU ARE TAKING REVLIMID
Things you must do:
FEMALE PATIENTS:
- Tell your doctor immediately if you become pregnant or suspect that you may be pregnant. You should also immediately stop taking Revlimid in this case.
ALL PATIENTS:
- Tell any other doctors, dentists, and pharmacists who are treating you that you are taking Revlimid.
- If you are about to be started on any new medicine, remind your doctor, dentist or pharmacist that you are taking Revlimid.
- Keep all of your doctor's appointments so that your progress can be checked.
Your doctor will do some blood tests regularly and will check your general condition to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do:
FEMALE PATIENTS:
- Do not become pregnant whilst taking Revlimid.
- Do not have sexual intercourse without using effective means of contraception described to you by your doctor.
MALE PATIENTS:
- Do not donate sperm during treatment or treatment interruption, or for at least 7 days after stopping treatment.
Revlimid can pass into human semen. - Do not have sexual intercourse without using effective means of contraception described to you by your doctor.
ALL PATIENTS:
- Do not donate blood during treatment or treatment interruption, or for at least 1 week after stopping treatment.
In Australia, patients with some types of cancer are permanently excluded from donating blood. - Do not stop taking Revlimid (unless you suspect that you are pregnant) or change the dose without first checking with your doctor.
- Do not let yourself run out of medicine over the weekend or on holidays.
- Do not give this medicine to anyone else, even if they have the same condition as you.
- Do not take this medicine to treat any other complaints unless your doctor or pharmacist tells you to.
- Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering.
In that case, return it to your pharmacist.
Things to be careful of:
Be careful driving or operating machinery until you know how Revlimid affects you. This medicine may cause dizziness, tiredness or blurred vision in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
SIDE EFFECTS
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Revlimid.
Like all medicines, Revlimid can have side effects, although not everybody gets them. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Tell your doctor if you notice any of the following and they worry you:
- Diarrhoea; constipation; feeling sick (also called nausea); vomiting; stomach pain; indigestion; dehydration; dry mouth; sore mouth: mouth ulcers: difficulty in speaking toothache; increase or decrease in weight; increase or decrease in appetite; loss of taste.
- Itchiness; rash; redness of the skin; dry skin; bruising; excessive sweating.
- Dizziness; fainting; headache; shaking or tremors; unusual weakness; night sweats; reduced sense of touch.
- Difficulty sleeping; depression; anxiety; feeling of confusion.
- Back pain; muscle spasms; muscle and/or joint pain; swollen joints; bone pain; muscular weakness; pain in the extremities; feeling tired; fall.
- Swelling of hands, ankles or feet.
The above list mainly includes the more common side effects of your medicine.
Tell your doctor immediately if you notice any of the following:
- Heart palpitations or fast heart beat, chest pains, dizziness or fainting, shortness of breath, weakness, or reduced ability to exercise.
These could be symptoms of atrial fibrillation (irregular heart beat) or tachycardia (fast heart beat). - Bleeding (including nose-bleeds) or bruising more easily than normal.
Revlimid can reduce the number of platelets, which are responsible for making the blood clot properly. Your doctor may monitor your blood cell numbers during treatment with Revlimid. - Tiredness, headaches, shortness of breath, dizziness and looking pale.
Revlimid can reduce the number of red blood cells that carry oxygen around the body. - Numbness, tingling, pins and needles or weakness of the arms and legs.
This may be due to nerve damage. - Blurred vision or difficulty seeing.
This could be due to a cataract in your eye(s). - Passing large amounts of urine, excessive thirst, and having a dry mouth and skin.
These could be symptoms of high blood sugar or diabetes. - Abnormal eye movements, convulsions, mood changes or irregular heart rhythms.
These could be due to low levels of minerals such as potassium, calcium, magnesium or sodium. - Tender swollen lymph nodes, low-grade fever, pain, or rash.
This could be due to worsening of your tumour (for patients with MCL).
The above list includes serious side effects that may require medical attention.
If any of the following happens, stop taking Revlimid and see a doctor immediately or go to Accident and Emergency at your nearest hospital:
- Shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, mouth, tongue or other parts of the body; rash, itching or hives on the skin.
These could be symptoms of an allergic reaction. - Severe blisters and bleeding in the lips, eyes, mouth, nose and genitals; painful red area on the skin that spreads quickly; peeling of the skin. You may have a high temperature, chills and muscle ache at the same time.
These could be due to rare but severe skin reactions such as Stevens-Johnson Syndrome, Toxic Epidermal Necrolysis and Drug Reaction with Eosinophilia and Systemic Symptoms. - Blurred vision; severe headache; weakness or numbness in the face, arm or leg; trouble speaking or understanding; loss of balance.
This may be due to a stroke which could be a result of blood clots in the blood vessels of your brain. - Sudden pain in your chest or difficulty in breathing.
This may be due to a heart attack or blood clots in the artery leading to your lungs. These blood clots can happen during treatment, or after treatment has stopped. - Chest pain, severe weakness, rapid or irregular heartbeat, and/or sudden, severe shortness of breath and coughing up pink, foamy mucus.
This could be due to heart failure, a condition where the heart muscle cannot pump blood strongly enough to supply blood throughout the body. - Pain or swelling in your legs, especially in your lower leg or calves.
This may be due to blood clots in the veins of your leg. These can happen during treatment, or after treatment has stopped. - Fever; severe chills; decreased urination; rapid pulse; rapid breathing; confusion; nausea; vomiting; diarrhoea; pain or burning when you urinate; hacking cough; phlegm; sore mouth or throat; flu-like symptoms; feeling of tension in the nose, cheeks and behind your eyes; or mouth ulcers.
These could symptoms of sepsis (blood infection) or other serious infections such as pneumonia. - Passing little or no urine; drowsiness; nausea; vomiting; or breathlessness.
These could be symptoms of kidney disease. - Abdominal pain, dark urine, fever, joint pain, loss of appetite, nausea and vomiting, yellowing of the skin and/or eyes.
These are symptoms of liver failure, which in some cases, may be due to Hepatitis B virus infection. Some cases of Hepatitis B virus infection may not result in symptoms initially.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor or pharmacist immediately if any of the side effects gets serious, or if you notice any other side effects not listed in this leaflet.
Other side effects not listed above may also occur in some people.
Some side effects (for example, changes in thyroid function, or blood pressure) can only be found when your doctor does tests from time to time to check your progress.
AFTER TAKING REVLIMID
Storage
Keep your capsules in a cool dry place where the temperature stays below 25°C.
Keep your capsules in the original package until it is time to take them.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, take any unused Revlimid capsules to your pharmacist.
Medicines should not be disposed of via wastewater or household waste. These measures will help to protect the environment.
PRODUCT DESCRIPTION
What Revlimid looks like:
The capsules are provided in packs. There are three (3) pack sizes available. A pack will contain either two blisters, each with seven capsules, giving a total of fourteen (14) capsules per pack; three blisters, each with seven capsules, giving a total of twenty-one (21) capsules per pack or four blisters, each with seven capsules, giving a total of twenty-eight (28) capsules per pack. Some strengths and pack sizes of Revlimid may not be available as not all strengths and pack sizes are being distributed.
Revlimid 2.5 mg capsules have a white body/blue-green opaque cap with "2.5 mg REV" written on them.
Revlimid 5 mg capsules are white to off-white opaque capsules with "5 mg REV" written on them.
Revlimid 7.5 mg capsules have a white body/pale yellow opaque cap with "7.5 mg REV" written on them.
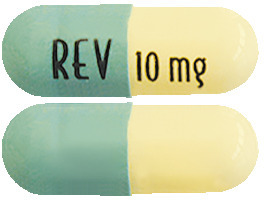
Revlimid 10 mg capsules are pale yellow opaque body/blue-green opaque cap capsules with "10 mg REV" written on them.
Revlimid 15 mg capsules are white to off-white opaque body / powder-blue opaque cap capsules with "15 mg REV" written on them.
Revlimid 20 mg capsules have a powder blue body/blue-green opaque cap with "20 mg REV" written on them.
Revlimid 25 mg capsules are white to off-white opaque capsules with "25 mg REV" written on them.
Ingredients
Revlimid capsules contain an active ingredient called lenalidomide.
The other ingredients are:
- lactose
- microcrystalline cellulose
- croscarmellose sodium, and
- magnesium stearate.
The capsule shells comprise of gelatin and titanium dioxide, and may also contain the following colourants:
- 2.5 mg capsules: indigo carmine [E132] and yellow iron oxide [E172]
- 7.5 mg capsules: yellow iron oxide [E172]
- 10 mg capsules: indigo carmine (E132) and yellow iron oxide (E172CI77492)
- 15 mg capsules; indigo carmine (E132)
- 20 mg capsules: indigo carmine [E132] and yellow iron oxide [E172].
The black printing ink contains shellac; ethanol; isopropyl alcohol; purified water; strong ammonia solution; potassium hydroxide; and black iron oxide (E172).
Distributor:
Revlimid is supplied in Australia by:
Celgene Pty Limited
Level 2, 4 Nexus Court
Mulgrave, VIC 3170
Telephone: 1800 CELGENE (1800 235 4363).
This leaflet was updated in April 2022.
Australian Registration Number:
Revlimid 2.5 mg AUST R 229850
Revlimid 5 mg AUST R 132510
Revlimid 7.5 mg AUST R 229851
Revlimid 10 mg AUST R 132514
Revlimid 15 mg AUST R 132515
Revlimid 20 mg AUST R 229852
Revlimid 25 mg AUST R 132516
® = Registered Trademark
(Celgene Version 8.0)
Published by MIMS May 2022



 Recommended dose adjustments for NDMM patients ineligible for transplant receiving Revlimid in combination with dexamethasone are found in Dose adjustments.
Recommended dose adjustments for NDMM patients ineligible for transplant receiving Revlimid in combination with dexamethasone are found in Dose adjustments. Recommended dose adjustments for NDMM post-transplant patients receiving Revlimid maintenance are found in Dose adjustments.
Recommended dose adjustments for NDMM post-transplant patients receiving Revlimid maintenance are found in Dose adjustments. Recommended dose adjustments for previously treated MM patients are found in Dose adjustments.
Recommended dose adjustments for previously treated MM patients are found in Dose adjustments. Recommended dose adjustments for MDS patients are found in Dose adjustments.
Recommended dose adjustments for MDS patients are found in Dose adjustments. Recommended dose adjustments for MCL patients are found in Dose adjustments.
Recommended dose adjustments for MCL patients are found in Dose adjustments. After initiation of Revlimid therapy, subsequent Revlimid dose modifications should be based on individual patient treatment tolerance. Patients with impaired renal function should be monitored for signs and symptoms of neutropenia or thrombocytopenia as per the recommendations (see Section 4.4 Special Warnings and Precautions for Use).
After initiation of Revlimid therapy, subsequent Revlimid dose modifications should be based on individual patient treatment tolerance. Patients with impaired renal function should be monitored for signs and symptoms of neutropenia or thrombocytopenia as per the recommendations (see Section 4.4 Special Warnings and Precautions for Use).


 If the dose of Revlimid was reduced for a haematologic dose-limiting toxicity (DLT), the dose of Revlimid may be re-increased to the next higher dose level (up to the starting dose) at the discretion of the treating physician if continued Revlimid/dexamethasone (Rev/dex) therapy resulted in improved bone marrow function (no DLT for at least 2 consecutive cycles and an ANC ≥ 1.5 x 109/L with a platelet count ≥ 100 x 109/L at the beginning of a new cycle at the current dose level).
If the dose of Revlimid was reduced for a haematologic dose-limiting toxicity (DLT), the dose of Revlimid may be re-increased to the next higher dose level (up to the starting dose) at the discretion of the treating physician if continued Revlimid/dexamethasone (Rev/dex) therapy resulted in improved bone marrow function (no DLT for at least 2 consecutive cycles and an ANC ≥ 1.5 x 109/L with a platelet count ≥ 100 x 109/L at the beginning of a new cycle at the current dose level).








 The following SAEs were also noted in both studies - lung infection, infection, urinary tract infection, herpes zoster and myelodysplastic syndrome.
The following SAEs were also noted in both studies - lung infection, infection, urinary tract infection, herpes zoster and myelodysplastic syndrome.
 The safety results (N = 148) from the phase 2 open label study MDS-003 are consistent with the findings from MDS-004. Neutropenia (66.2%) and thrombocytopenia (64.9%) were the most frequently reported AEs, followed by diarrhoea (60.1%), pruritus (44.6%), fatigue (42.6%), rash (37.8%) and arthralgia (31.8%).
The safety results (N = 148) from the phase 2 open label study MDS-003 are consistent with the findings from MDS-004. Neutropenia (66.2%) and thrombocytopenia (64.9%) were the most frequently reported AEs, followed by diarrhoea (60.1%), pruritus (44.6%), fatigue (42.6%), rash (37.8%) and arthralgia (31.8%).

 The primary efficacy endpoint in the study was progression free survival (PFS). The demographics and disease-related baseline characteristics of the patients were similar across the two treatment groups and reflected a broad NDMM patient population. In total 523 patients were enrolled into the study, with 263 patients randomised to Rbd and 260 patients randomised to Rd, 162 (30.9%) subjects were stratified as "no intent to transplant at disease progression", 81 subjects in the Rbd arm and 81 subjects in the Rd arm.
The primary efficacy endpoint in the study was progression free survival (PFS). The demographics and disease-related baseline characteristics of the patients were similar across the two treatment groups and reflected a broad NDMM patient population. In total 523 patients were enrolled into the study, with 263 patients randomised to Rbd and 260 patients randomised to Rd, 162 (30.9%) subjects were stratified as "no intent to transplant at disease progression", 81 subjects in the Rbd arm and 81 subjects in the Rd arm.


 Study IFM 2005-02 recruited patients aged < 65 years at diagnosis who had undergone treatment with high-dose chemotherapy supported by ASCT and had achieved at least a stable disease response at the time of haematologic recovery.
Study IFM 2005-02 recruited patients aged < 65 years at diagnosis who had undergone treatment with high-dose chemotherapy supported by ASCT and had achieved at least a stable disease response at the time of haematologic recovery. The efficacy of Revlimid maintenance versus placebo/no maintenance as a treatment for adult NDMM patients who have undergone ASCT, as measured by overall survival (OS), was further assessed in a meta-analysis of 3 randomised controlled trials (including Studies CALGB 100104, IFM 2005-02 and GIMEMA). A total of 1209 patients are included in the meta-analysis. The demographic and disease characteristics were reflective of a typical transplant-eligible patient population with NDMM.
The efficacy of Revlimid maintenance versus placebo/no maintenance as a treatment for adult NDMM patients who have undergone ASCT, as measured by overall survival (OS), was further assessed in a meta-analysis of 3 randomised controlled trials (including Studies CALGB 100104, IFM 2005-02 and GIMEMA). A total of 1209 patients are included in the meta-analysis. The demographic and disease characteristics were reflective of a typical transplant-eligible patient population with NDMM.




