What is in this leaflet
This leaflet answers some common questions about RiaSTAP®.
It does not contain all the available information. If you require further information about this medicine or your treatment generally, or if you have any questions or are not sure about something in this leaflet, consult your doctor.
All medicines have benefits and risks. Your doctor has weighed the benefits that RiaSTAP® will have for you against the possible risks.
If you have any concerns about taking this medicine, ask your doctor. Follow your doctor’s advice even if it is different from what this leaflet says.
Keep this leaflet with the medicine. You may need to read it again.
The information in this leaflet is subject to change.
Please check with your doctor whether there is any new information about this medicine that you should know since you were last treated.
What RiaSTAP® is used for
This medicine is used for the treatment of acute bleeding in people with an absence or low level of human fibrinogen (congenital lack of fibrinogen).
Ask your doctor if you have any questions about why RiaSTAP® has been prescribed for you.
How RiaSTAP® works
This product is made from human plasma (this is the liquid part of the blood). It contains human fibrinogen as the active ingredient. Human fibrinogen is a protein which is important for blood clotting (coagulation). If you have missing or malfunctioning fibrinogen the blood does not clot as quickly as it should which results in an increased tendency of bleeding. The replacement of human fibrinogen with RiaSTAP® will repair the coagulation mechanisms.
Before you are given RiaSTAP®
When you must not have it
Do not have RiaSTAP® if you are allergic to:
- human fibrinogen
- any of the ingredients listed at the end of this leaflet.
If you are not sure whether you should be given this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you:
- have had a heart attack (a history of coronary heart disease or myocardial infarction)
- have just had surgery
- suffer from liver disease
- will have surgery soon
- are more likely to suffer from blood clots than normal.
Tell your doctor if you are pregnant or breast feeding. This medicine should only be used if clearly needed during pregnancy or breast feeding.
Tell your doctor if you are on a controlled sodium diet. This medicine contains sodium which should be taken into consideration.
Tell your doctor if you are allergic to any medicine or food.
If you have not told your doctor about any of the above, tell them before you are given RiaSTAP®. Your doctor can discuss with you the risks and benefits involved with using this medicine.
About blood products
This product is made from human plasma (this is the liquid part of blood). When products are made from human blood or plasma and injected into you, it is possible that viruses or other substances could be present in the product and cause an illness. These could be viruses such as hepatitis, human immunodeficiency virus (HIV), or parvovirus B19. There could also be other infectious agents some of which may not yet have been discovered.
To reduce the risk of infection extra steps are taken when manufacturing this product. Strict controls are applied when selecting blood donors and donations. The product is specially treated to kill and remove viruses. These special treatments are considered effective against certain viruses known as enveloped viruses (such as HIV and hepatitis B and C) and also for the non-enveloped virus hepatitis A. The measures taken may be of limited value against non-enveloped viruses such as parvovirus B19. Despite these measures, the risk of transmitting infection cannot be totally eliminated.
Vaccines are available against some of these viruses and your doctor will be able to help you decide whether you should have any of those vaccines.
Please discuss the risks and benefits of this medicine with your doctor.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may affect the way other medicines work.
How to use RiaSTAP®
Treatment should be started and supervised by a doctor. RiaSTAP® is reconstituted with 50 mL of Water for Injections and then administered by slow intravenous injection at a rate not exceeding 5 mL per minute.
The following procedures are given as a guide only:
Preparing RiaSTAP® for reconstitution
- Ensure you have all the required components to reconstitute RiaSTAP®.
- Allow the vials of RiaSTAP® and Water for Injections (WFI) to reach room temperature prior to use, which may take up to one hour. Do not warm the WFI in hot water.
- Remove jewellery, watches, rings, etc.
- Wash hands with soap and water, dry with a clean towel.
- Select an appropriate work area with good lighting and a surface which can be cleaned.
- Using a clean cloth or paper towel, clean the preparation area with methylated spirits.
- Gather the components to be used (supplied in the RiaSTAP® carton).
Components
- One vial of RiaSTAP®
- One vial of 50 mL Water for injections
- One transfer set
- One dispensing pin
- One syringe filter.
Check the expiry date of each item. Do not use if expired.
Instructions for RiaSTAP® reconstitution
Follow these steps to prepare the injection.
- Wash hands with soap and water, dry with a clean towel or use gloves before reconstituting the product.
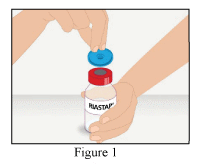
- Remove the cap from the RiaSTAP® vial to expose the central portion of the rubber stopper (Figure 1).
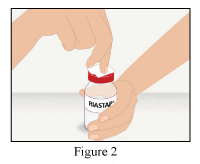
- Clean the surface of the rubber stopper with an antiseptic solution and allow to dry (Figure 2).
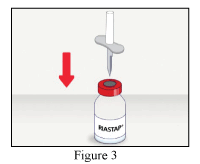
- Remove the safety cap from one end of the provided transfer set and pierce the stopper of the RiaSTAP® vial (Figure 3).
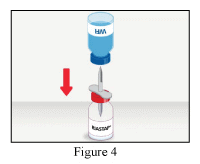
- Remove the safety cap from the other end of the transfer set, invert the WFI vial, apply gentle pressure to pierce the stopper and transfer the contents into the RiaSTAP® vial (Figure 4).
- Discard the WFI vial and remove the transfer set from the RiaSTAP® vial.
- Gently swirl the RiaSTAP® vial to ensure the product is fully dissolved (Figure 5).
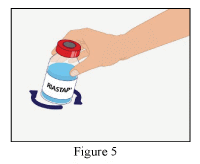
- Avoid shaking which causes formation of foam.
- The powder should be completely reconstituted within 15 minutes (generally 5 to 10 minutes)
- Open the plastic blister containing the dispensing pin (Figure 6).
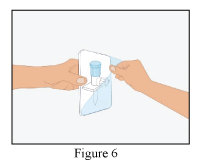
- Take the provided dispensing pin and insert into the stopper of the vial with the reconstituted RiaSTAP® (Figure 7).
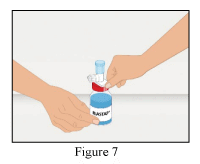
- After the dispensing pin is inserted, remove the cap. After the cap is removed, do not touch the exposed surface.
- Open the blister with the provided syringe filter (Figure 8).
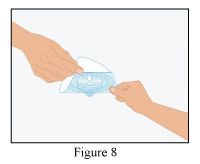
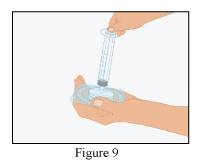
- Screw the syringe (not supplied) onto the filter (Figure 9).
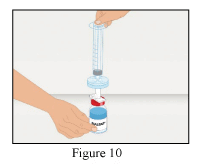
- Screw the syringe with the mounted filter onto the dispensing pin (Figure 10).
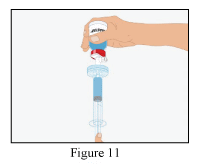
- Draw the reconstituted RiaSTAP® into the syringe (Figure 11).
- When completed, remove the syringe filter, dispensing pin and empty vial from the syringe, dispose of appropriately, and proceed with administration
After reconstitution the solution should be clear or slightly opalescent, that is it might sparkle when held up to the light but must not contain any obvious particles.
Do not mix RiaSTAP® with other medicinal products or diluents either before or during administration.
If it is not injected immediately it must be stored below 25°C and used within 6 hours of reconstitution. The reconstituted solution should not be stored in the refrigerator.
Any unused portion remaining in the vial must be discarded appropriately.
Every time you are given RiaSTAP®, the date of injection, the batch number and the injected volume should be recorded.
How much is given
The dose and duration of administration of RiaSTAP® depends on the severity of your fibrinogen deficiency and the location and extent of your bleeding.
As some patients may have an increased risk of blood clotting (thrombosis) the use of RiaSTAP® should only be given by doctors experienced in this treatment.
When it is given
Your doctor will discuss with you when you should be given RiaSTAP®.
If too much is given (overdose)
Overdose may enhance the risk of blood clots (thrombosis). Your doctor should regularly check your clotting status during your treatment.
If you have any questions consult your doctor.
While you are having RiaSTAP®
Things you must do
If you notice signs or symptoms of a serious allergy or anaphylaxis (see Side effects) while you are being given RiaSTAP® tell your doctor immediately as the administration of RiaSTAP® should be stopped immediately.
Things you must not do
Do not give or share your medicine with anyone else, even if they have the same condition as you.
Use this medicine in one patient on one occasion only.
Side effects
Tell your doctor as soon as possible if you do not feel well while you are being given RiaSTAP®.
This medicine helps most people with congenital fibrinogen deficiency who suffer a bleeding episode, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Tell your doctor immediately if you notice any of the following symptoms which may be signs of allergy as the injection of RiaSTAP® should be stopped:
- feeling faint (fall in blood pressure)
- reddening of the skin
- rash
- difficulty in breathing
- dizziness
- itching
- shortness of breath.
Tell your doctor if you notice the following and it worries you:
- fever.
This is the most common side effect of RiaSTAP®.
There is a risk of blood clots forming in blood vessels (thrombosis) particularly when high or repeated doses are given. The use of RiaSTAP® should only be given by experienced doctors.
Other side effects not listed above may also occur in some patients. Tell your doctor if you notice any other effects.
Do not be alarmed by this list of possible side effects.
This medicine does not generally cause any undesired reactions.
How to store RiaSTAP®
Store at 2°C to 8°C (Refrigerate. Do not freeze). Keep the product in the carton in order to protect it from light.
Do not use after the expiry date.
Keep it out of the sight and reach of children.
Product description
What it looks like
RiaSTAP® is a white powder contained in a glass vial.
RiaSTAP® comes in a carton which also contains:
- one vial of 50 mL Water for Injections
- one transfer set
- one dispensing pin
- one syringe filter.
After reconstitution the solution should be clear or slightly opalescent, that is it might sparkle when held up to the light but must not contain any obvious particles.
Ingredients
RiaSTAP® contains 1 gram of human fibrinogen as the active ingredient.
It also contains:
- albumin
- arginine hydrochloride
- sodium chloride
- sodium citrate.
Sponsor
CSL Behring (Australia) Pty Ltd
ABN 48 160 734 761
189-209 Camp Road
Broadmeadows VIC 3047
Australia
Manufacturer
CSL Behring GmbH
35041 Marburg
Germany
Date of revision
May 2020
Australian Register Number: AUST R 162828
® Registered trademark of CSL Limited Group of Companies
Published by MIMS July 2020


 For safety with respect to transmissible agents, see Section 4.4 Special Warnings and Precautions for Use, Pathogen safety.
For safety with respect to transmissible agents, see Section 4.4 Special Warnings and Precautions for Use, Pathogen safety. Adverse reactions encountered during the clinical trials are outlined, see Section 4.8 Adverse Effects (Undesirable Effects).
Adverse reactions encountered during the clinical trials are outlined, see Section 4.8 Adverse Effects (Undesirable Effects).
