WHAT IS IN THIS LEAFLET
This leaflet answers some common questions about Risedronate Sandoz.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
WHAT RISEDRONATE SANDOZ IS USED FOR
Risedronate Sandoz is used to treat the following bone diseases:
- osteoporosis (brittle or fragile bones that may fracture easily)
- osteoporosis caused by taking steroids.
It contains the active ingredient Risedronate sodium. Risedronate sodium belongs to a group of medicines called biphosphonates.
It works directly on your bones to make them stronger and therefore less likely to break or fracture, by slowing down the process of old bone being removed and helping to rebuild bone mass.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
There is no evidence that Risedronate Sandoz is addictive.
Risedronate Sandoz is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children under the age of 18 years.
BEFORE YOU TAKE RISEDRONATE SANDOZ
When you must not take it
Do not take this medicine if you have an allergy to:
- risedronate sodium, the active ingredient, or to any of the other ingredients listed at the end of this leaflet under Product Description.
- any other similar medicines, such as alendronate, pamidronate or any other bisphosphonates.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take this medicine if you:
- are unable to stand or sit upright for at least 30 minutes
- have a condition called hypocalcaemia (a low level of calcium in the blood)
- have severe kidney problems.
Do not take Risedronate Sandoz if you are pregnant. Risedronate Sandoz is not recommended for use during pregnancy, unless you and your doctor have discussed the risks and benefits involved.
Do not take Risedronate Sandoz if you are breastfeeding. It is not known whether Risedronate Sandoz passes into breast milk.
Do not take Risedronate Sandoz after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If you take this medicine after the expiry date has passed, it may not work as well.
Talk to your doctor or pharmacist if you are not sure whether you should start taking Risedronate Sandoz.
Before you start to take it
Tell your doctor or pharmacist if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you are pregnant or intend to become pregnant. Your doctor will discuss the possible risks and benefits of using Risedronate Sandoz during pregnancy.
Tell your doctor or pharmacist if you are breastfeeding or plan to breastfeed.
Tell your doctor if you have or have had any of the following medical conditions:
- disturbances of bone and mineral metabolism (for example vitamin D deficiency, parathyroid hormone abnormalities)
- problems with the tube that takes food from your mouth to your stomach (oesophagus), such as ulcers
- pain, swelling or numbness of the jaw or a “heavy jaw feeling” or loosening of a tooth.
Check with your doctor or dentist to see if a dental check-up is required before starting Risedronate Sandoz. This is especially important if you are receiving medicines or therapy used to treat cancer or taking corticosteroids, such as prednisone or cortisone.
Tell your doctor about any of the above, before you start taking Risedronate Sandoz.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Risedronate Sandoz may interfere with each other if taken at the same time. These include:
- antacids, medicines used to treat indigestion e.g. Gaviscon®, Mylanta®
- other products containing calcium
- iron supplements.
Check with your doctor or pharmacist if you are not sure whether you are taking any of these products.
You may need to stop taking these products or take them at a different time of day to Risedronate Sandoz.
You can take aspirin while you are being treated with Risedronate Sandoz.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking Risedronate Sandoz.
HOW TO TAKE RISEDRONATE SANDOZ
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
How much to take
The standard dose for this medicine is one tablet once a week.
Ask your doctor or pharmacist if you are unsure of the correct dose for you. They will tell you exactly how much to take.
How to take it
Take Risedronate Sandoz in the morning, at least 30 minutes before your first meal, drink or medication of the day. Risedronate Sandoz is most effective when your stomach is empty.
Swallow one tablet whole with a glass of plain water. Do not chew or suck the tablet. It is important to take Risedronate Sandoz with plain water only (120 mL), not mineral water. Mineral water and other drinks, including fruit juices, coffee and tea, will reduce the effect of Risedronate Sandoz.
Take the tablet while sitting or standing upright. Do not lie down immediately after swallowing it.
Stay upright for at least 30 minutes after swallowing Risedronate Sandoz and do not take any food, medicines or drinks other than plain tap water during this time. It is important to stay upright (sitting, standing or walking around) for at least 30 minutes after swallowing your tablet. It is also very important to stay upright until after you have eaten your first food of the day. This will help make sure the tablet reaches your stomach quickly and helps avoid irritation to your oesophagus.
When to take Risedronate Sandoz
Take Risedronate Sandoz in the morning, 30 minutes to 1 hour before your first meal, drink or medication of the day.
Take your Risedronate Sandoz on the same day each week. This tablet should be taken each week. Choose a day of the week that suits you the best.
How long to take Risedronate Sandoz
Continue taking your medicine for as long as your doctor recommends it.
Do not stop taking Risedronate Sandoz without checking with your doctor or pharmacist.
If you forget to take it
If you have forgotten to take this medicine, just take it on the day you remember.
Do not take two tablets in one day to make up for the tablet you missed. Return to taking one tablet once a week, as originally scheduled on your chosen day.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26 or New Zealand 0800 POISON or 0800 764766) for advice, or go to Accident and Emergency at the nearest hospital.
If you or somebody else has accidentally taken a large number of tablets, drink a full glass of milk or antacids. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
WHILE YOU ARE TAKING RISEDRONATE SANDOZ
Things you must do
Tell any other doctors, dentists, and pharmacists who are treating you that you are taking Risedronate Sandoz.
If you require a dental procedure, tell your dentist that you are taking Risedronate Sandoz. Invasive dental procedures should be avoided where possible. This type of medicine may cause jaw-bone problems in some people. Jaw-bone problems may include infection, and delayed healing after teeth are pulled out or other work that involves drilling into the jaw.
If you develop a toothache, jaw pain, painful exposed bone or swelling, especially following dental work, tell your doctor or dentist immediately.
Speak to your doctor and dentist about good oral hygiene and regular dental check-ups while you are using Risedronate Sandoz.
If you are about to be started on any new medicine, tell your doctor, dentist or pharmacist that you are taking Risedronate Sandoz.
If you become pregnant while taking Risedronate Sandoz tell your doctor or pharmacist.
If you develop new or unusual pain in your thigh or hip, tell your doctor.
Rarely, patients have experienced fracture in a specifc part of the thigh bone..
Things you must not do
Do not lie down for 30 minutes after taking Risedronate Sandoz. Do not have any food or drink, except for plain water for 30 minutes after taking Risedronate Sandoz.
Do not take Risedronate Sandoz to treat any other complaints unless your doctor tells you to.
Do not give Risedronate Sandoz to anyone else, even if they have the same condition as you.
Do not stop taking Risedronate Sandoz without checking with your doctor or pharmacist.
SIDE EFFECTS
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Risedronate Sandoz. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- stomach pain
- diarrhoea
- aching muscles, joints or bones
- headache
- nausea
- runny nose
- sore throat
- dizziness
These side effects are usually mild.
Tell your doctor immediately if you notice any of the following:
- skin rash or redness of the skin, sometimes made worse by sunlight, itchiness
- blurred vision, pain or redness in the eyes
- problems with your jaw or teeth, associated with delayed healing and/or infection often following a tooth extraction or invasive dental work
These side effects are rare.
If any of the following happen, stop taking Risedronate Sandoz and tell your doctor immediately:
- difficulty or pain on swallowing
- chest pain
- new or worsening heartburn.
These side effects may be due to irritation or ulceration of the food pipe. They may worsen if you continue taking the tablets. These side effects are rare.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- swelling of the face, lips, mouth, throat or tongue
- severe skin reactions, hair loss.
These may be serious side effects. You may need urgent medical attention.
Other side effects not listed above may occur in some patients.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell.
AFTER USING RISEDRONATE SANDOZ
Storage
Keep your medicine in the original container.
If you take the tablets out of the original container they may not keep well.
Keep your medicine in a cool dry place where the temperature stays below 25°C.
Do not store Risedronate Sandoz or any other medicine in the bathroom or near a sink.
Do not leave medicines on a window sill or in the car on hot days. Heat and dampness can destroy some medicines.
Keep medicines where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicines that are left over.
PRODUCT DESCRIPTION
What it looks like
Risedronate Sandoz 35mg - orange, oval, biconvex film-coated tablet marked 35 on one side.
Available in blister packs of 4 tablets.
Ingredients
Active ingredient:
Risedronate Sandoz 35mg - 35mg risedronate sodium per tablet
Inactive ingredients:
- lactose monohydrate
- crospovidone
- microcrystalline cellulose
- magnesium stearate
- Opadry Orange (containing hypromellose, titanium dioxide, macrogol 400, iron oxide yellow, iron oxide red).
This medicine does not contain sucrose, gluten, tartrazine or any other azo dyes.
Supplier/Distributor
Sandoz Pty Ltd
ABN 60 075 449 553
54 Waterloo Road
Macquarie Park, NSW 2113
Australia
Tel: 1800 726 369
Novartis New Zealand Limited
PO Box 99102
Newmarket, Auckland 1149
New Zealand
Tel: 0800 354 335
This leaflet was prepared in March 2019.
Australian Register Number
Risedronate Sandoz 35mg film-coated tablets: AUST R 154854
Published by MIMS May 2019
 In a two year study in men with osteoporosis, the overall safety and tolerability were similar between the treatment and the placebo groups. Adverse experiences were consistent with those previously observed in women.
In a two year study in men with osteoporosis, the overall safety and tolerability were similar between the treatment and the placebo groups. Adverse experiences were consistent with those previously observed in women.
 The incidence of nonvertebral fractures in the pooled analysis (RVN and RVE) was lower in the risedronate 5 mg group than in the placebo group for all fractures at these sites combined, as well as for the wrist, humerus, pelvis and leg separately. This difference was significant for all nonvertebral osteoporosis related fractures (p = 0.005), as well as for the humerus (p = 0.024) and pelvis (p = 0.044), while a trend was seen at the wrist (p = 0.075) (see Table 2).
The incidence of nonvertebral fractures in the pooled analysis (RVN and RVE) was lower in the risedronate 5 mg group than in the placebo group for all fractures at these sites combined, as well as for the wrist, humerus, pelvis and leg separately. This difference was significant for all nonvertebral osteoporosis related fractures (p = 0.005), as well as for the humerus (p = 0.024) and pelvis (p = 0.044), while a trend was seen at the wrist (p = 0.075) (see Table 2).

 Very few patients in any treatment group had new fractured vertebrae at month 12 (5 mg daily: 1.5%; 35 mg once a week: 1.3%). No patient had more than one new fractured vertebra. There were no statistically significant differences in the percentage of patients with new vertebral fractures among the two treatment groups.
Very few patients in any treatment group had new fractured vertebrae at month 12 (5 mg daily: 1.5%; 35 mg once a week: 1.3%). No patient had more than one new fractured vertebra. There were no statistically significant differences in the percentage of patients with new vertebral fractures among the two treatment groups. A second study of similar design enrolled 290 patients with continuing, long-term use (greater than or equal to six months) of corticosteroids for rheumatic, skin and pulmonary diseases. The baseline mean lumbar spine BMD was low (1.64 SD below the young healthy population mean), with 28% of the patients more than 2.5 SD below the mean. All patients in this study received supplemental calcium 1,000 mg/day. Patients also received supplemental vitamin D 400 IU/day. After one year of treatment, the BMD of the placebo group remained near baseline levels at the lumbar spine, femoral neck and trochanter. Risedronate 5 mg once daily improved bone mass with a statistically significant mean increase compared to placebo of 2.7% at the lumbar spine and 1.9% at the femoral neck as shown in Figure 5. At the trochanter, a statistically significant increase from baseline was demonstrated (2.4%). Risedronate was effective regardless of age, race, gender, underlying disease, corticosteroid dose or baseline BMD. See Figure 5.
A second study of similar design enrolled 290 patients with continuing, long-term use (greater than or equal to six months) of corticosteroids for rheumatic, skin and pulmonary diseases. The baseline mean lumbar spine BMD was low (1.64 SD below the young healthy population mean), with 28% of the patients more than 2.5 SD below the mean. All patients in this study received supplemental calcium 1,000 mg/day. Patients also received supplemental vitamin D 400 IU/day. After one year of treatment, the BMD of the placebo group remained near baseline levels at the lumbar spine, femoral neck and trochanter. Risedronate 5 mg once daily improved bone mass with a statistically significant mean increase compared to placebo of 2.7% at the lumbar spine and 1.9% at the femoral neck as shown in Figure 5. At the trochanter, a statistically significant increase from baseline was demonstrated (2.4%). Risedronate was effective regardless of age, race, gender, underlying disease, corticosteroid dose or baseline BMD. See Figure 5.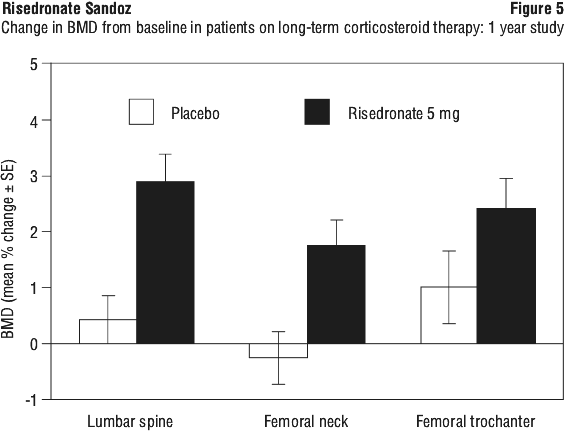
 Chemical formula: [1-hydroxy-2-(3-pyridinyl)ethylidene]bis (phosphonic acid) monosodium salt.
Chemical formula: [1-hydroxy-2-(3-pyridinyl)ethylidene]bis (phosphonic acid) monosodium salt.