Notes
Distributed by Medsurge Healthcare Pty Ltd
1 Name of Medicine
Prucalopride.
2 Qualitative and Quantitative Composition
Each film-coated tablet contains 1 mg or 2 mg of prucalopride as the succinate salt.
Excipient of known effect.
Sugars as lactose.
For a full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Film-coated tablet.
1 mg.
White to off-white, round, tablets with "C" debossed on one side and "11" on the other side.
2 mg.
Pink, round, tablets with "C" debossed on one side and "12" on the other side.4.1 Therapeutic Indications
Prucalopride is indicated for the treatment of chronic functional constipation in adults in whom laxatives fail to provide adequate relief.
Before prucalopride is considered patients must have tried at least two different types of laxatives from different classes (at the highest tolerated recommended doses) for at least six months, but have not had adequate relief from constipation.
If treatment with prucalopride is not effective within four weeks, the benefit of continuing treatment should be reconsidered.
4.2 Dose and Method of Administration
Dosage.
Prucalopride film-coated tablets are for oral use and can be taken with or without food.
Adults.
2 mg once daily.
Method of administration.
Prucalopride film-coated tablets are for oral use and can be taken with or without food.
Dosage adjustment.
Hepatic impairment.
The dose for patients with severe hepatic impairment (Child-Pugh class C) is 1 mg once daily (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties). No dose adjustment is required for patients with mild to moderate hepatic impairment.
Due to the specific mode of action of prucalopride (stimulation of propulsive motility), exceeding the daily dose of 2 mg is not expected to increase efficacy.
Renal impairment.
The dose for patients with severe renal impairment not requiring dialysis (GFR < 30 mL/min/1.73 m2) is 1 mg once daily (see Section 4.3 Contraindications; Section 5.2 Pharmacokinetic Properties). No dose adjustment is required for patients with mild to moderate renal impairment.
Elderly (> 65 years).
Start with one 1 mg tablet once daily (see Section 5.2 Pharmacokinetic Properties); if needed the dose can be increased to 2 mg once daily.
Paediatric.
Prucalopride is not recommended in children and adolescents younger than 18 years.
Treatment duration.
If the intake of the prescribed once daily dose of prucalopride is not effective after four weeks of treatment, the patient should be re-examined and the benefit of continuing treatment should be reconsidered.
Efficacy and safety of prucalopride has been established in double-blind placebo-controlled studies for up to 12 weeks. Patients should be reassessed after 12 weeks prior to continuation of treatment with prucalopride.
Use with laxatives.
Efficacy and safety of prucalopride when used in combination with laxatives has not been assessed, although laxatives were used as rescue medications in the pivotal clinical trials.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients.
Renal impairment requiring dialysis.
Intestinal perforation or obstruction due to structural or functional disorder of the gut wall, obstructive ileus and active severe inflammatory conditions of the intestinal tract, such as Crohn's disease, and ulcerative colitis and toxic megacolon/megarectum.
Recent bowel surgery.
4.4 Special Warnings and Precautions for Use
Prior to receiving prucalopride patients require a thorough history and examination to exclude secondary causes of constipation and to establish failure to respond adequately to at least 2 different types of laxatives from different classes for at least 6 months.
The safety and efficacy of prucalopride in combination with laxatives has not been assessed, although laxatives were used as rescue medications in the pivotal clinical trials.
Secondary constipation.
Efficacy and safety of prucalopride has been demonstrated only in patients with chronic functional constipation. Efficacy and safety of prucalopride in patients with secondary causes of constipation including endocrine disorders, metabolic disorders and neurologic disorders have not been assessed and use in these patients is not recommended. Efficacy and safety of prucalopride in patients with medication-related constipation, including constipation due to opioid use as a secondary cause of constipation, has not been demonstrated and use of prucalopride is not recommended.
Concomitant disease.
There is limited information in patients with severe and clinically unstable concomitant disease (e.g. liver, cardiovascular or lung disease, neurological or psychiatric disorders, cancer or AIDS and other endocrine disorders). Therefore, caution should be exercised when prescribing prucalopride to patients with these conditions. In particular, prucalopride should be used with caution in patients with a history of arrhythmias or ischaemic cardiovascular disease.
Oral contraceptives.
In case of severe diarrhoea, the efficacy of oral contraceptives may be reduced and the use of an additional contraceptive method is recommended to prevent possible failure of oral contraception (see the prescribing information of the oral contraceptive).
Lactose.
The tablets contain lactose monohydrate. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption must not take this medicinal product.
Psychiatric disorders.
Suicides, suicide attempts, and suicidal ideation have been reported in clinical trials and post- market experience. A causal association between treatment with prucalopride and suicidal ideation and behaviour has not been established. Patients should be monitored for persistent worsening of depression or the emergence of suicidal thoughts and behaviours. Counsel the patients and their caregivers and family members to be aware of any unusual changes in mood or behaviour, and to discontinue prucalopride and alert the healthcare provider immediately.
Use in hepatic impairment.
A lower dose is recommended for patients with severe hepatic impairment (see Section 4.2 Dose and Method of Administration).
Use in renal impairment.
Renal excretion is the main route of elimination of prucalopride (see Section 5.2 Pharmacokinetic Properties). A dose of 1 mg is recommended in subjects with severe renal impairment (see Section 4.2 Dose and Method of Administration).
Use in the elderly.
Start with one 1 mg tablet once daily (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties). If needed, the dose can be increased to 2 mg once daily.
Paediatric use.
Prucalopride is not recommended in children and adolescents younger than 18 years.
Effects on laboratory tests.
No effects are known.4.5 Interactions with Other Medicines and Other Forms of Interactions
Potentiation of effect.
Ketoconazole (200 mg b.i.d.), a potent inhibitor of CYP3A4 and of P-gp, increased the area under the curve (AUC) of prucalopride by approximately 40%. This effect is too small to be clinically relevant and is likely attributable to inhibition of P-gp mediated renal transport. Interactions of similar magnitude as observed with ketoconazole may also occur with other potent inhibitors of P-gp such as verapamil, cyclosporine A and quinidine. Prucalopride is likely also secreted via another renal transporter(s). Inhibition of all transporters involved in the active secretion of prucalopride (including P-gp) may theoretically increase the exposure by up to 75%.
Reduction of effect.
Because of the mechanism of action, the use of atropine-like substances may reduce the 5-HT4 receptor mediated effects of prucalopride.
In vitro data indicate that prucalopride has a low interaction potential, and therapeutic concentrations of prucalopride are not expected to affect the CYP-mediated metabolism of co-medicated medicinal products. Prucalopride is a weak substrate for P-glycoprotein (P-gp). Prucalopride is a weak in vitro inhibitor of P-gp and BCRP transporters, and it is not a significant inhibitor of OATP1B1, OATP1B3, OAT1, OAT3, BSEP and MRP2 transporters.
Studies in healthy subjects showed that there were no clinically relevant effects of prucalopride on the pharmacokinetics of warfarin, digoxin, alcohol, paroxetine and oral contraceptives. A 30% increase in the plasma concentrations of erythromycin was found during prucalopride co- treatment. The mechanism for this interaction is not fully known, but the available data support that this is the consequence of the high intrinsic variability in erythromycin kinetics, rather than a direct effect of prucalopride.
There are no data on the effect of prucalopride on SSRIs other than paroxetine or of the effect of SSRIs on prucalopride.
Therapeutic doses of probenecid, cimetidine, erythromycin and paroxetine did not affect the pharmacokinetics of prucalopride.
Prucalopride should be used with caution in patients receiving concomitant drugs known to cause QTc prolongation.
Interactions with food have not been observed.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There is no information on the effects of prucalopride on human fertility. There were no adverse effects on the fertility of rats treated orally or subcutaneously with prucalopride at doses up to 20 mg/kg/day, with estimated exposure about 100 times clinical exposure at the MRHD, based on AUC.
(Category B1)
Experience with prucalopride during pregnancy is limited. Cases of spontaneous abortion have been observed during clinical studies, although, in the presence of other risk factors, the relationship to prucalopride is unknown. Prucalopride is not recommended during pregnancy, and women of childbearing potential should use effective contraception during treatment with prucalopride.
There was no evidence of teratogenicity in rats or rabbits treated with prucalopride during the period of organogenesis at oral doses up to 80 mg/kg/day, (respective exposures about 400 times and 40 times the clinical exposure at the MRHD, based on AUC).
Prucalopride is excreted in breast milk. However, at therapeutic doses of prucalopride, no effects on the breastfed newborns/infants are anticipated. In the absence of human data in women who breastfed while taking prucalopride, it is not recommended to use prucalopride during breastfeeding.
Oral administration of prucalopride to rats from early gestation to weaning at doses up to 80 mg/kg/day was associated with slightly reduced pup survival due to maternotoxicity, with estimated exposure at least 100 times clinical exposure at the MRHD, based on AUC.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects of prucalopride on the ability to drive and use machines have been performed. Prucalopride has been associated with dizziness and fatigue particularly during the first day of treatment which may have an effect on driving and using machines (see Section 4.8 Adverse Effects (Undesirable Effects)).
4.8 Adverse Effects (Undesirable Effects)
Prucalopride has been given orally to approximately 2,700 patients with chronic constipation in controlled clinical studies. Of these patients, almost 1,000 patients received prucalopride at the recommended dose of 2 mg per day, while about 1.300 patients were treated with 4 mg prucalopride daily. Total exposure in the clinical development plan exceeded 2,600 patient years. The most frequently reported adverse reactions associated with prucalopride therapy are headache and gastrointestinal symptoms (abdominal pain, nausea or diarrhoea) occurring in approximately 20% of patients each. The adverse reactions occur predominantly at the start of therapy and usually disappear within a few days with continued treatment. Other adverse reactions have been reported occasionally. The majority of adverse events were mild to moderate in intensity.
Adverse events reported by more than 2.0% of the patients in the "All prucalopride" treatment group in the phase II and III double-blind placebo-controlled trials in patients with chronic constipation are shown in Table 1.
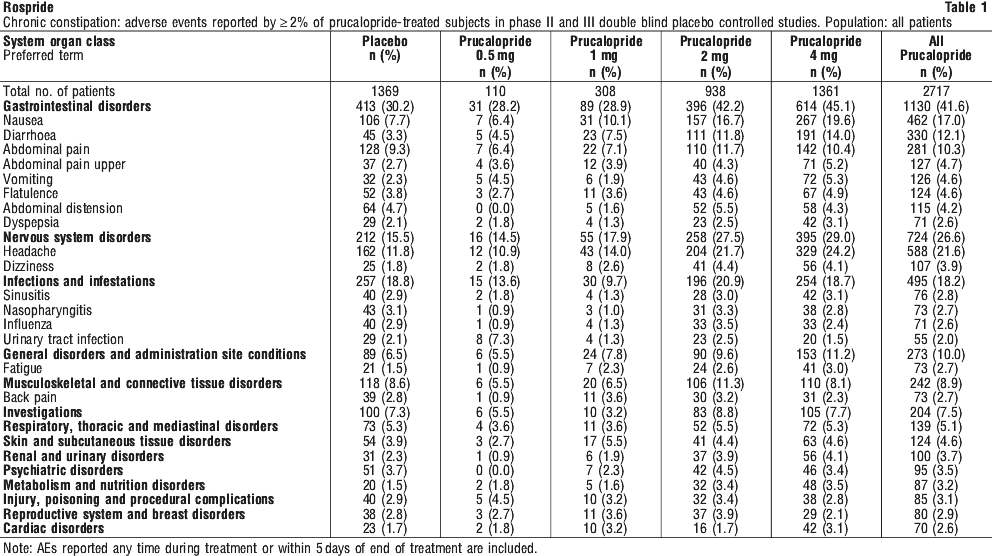 A total of 564 elderly patients (≥ 65 years) with chronic constipation were treated with prucalopride in double-blind studies, with a total exposure of 63 person-years. Most patients in the phase II/III double-blind placebo-controlled studies were younger than 65 years. The incidence of adverse events in the < 65 years old group was 71.2% (1534 out of 2153 patients) in the prucalopride group, and 61.6% (712 out of 1155) in the placebo group. In the group of patients older than 65 years, the incidence of adverse events in the prucalopride group was 58.7% (331 out of 564) and in the placebo group 52.8% (113 out of 214). Similar to the younger age group, the most common adverse events with prucalopride treatment among the elderly (> 65 years) groups were gastrointestinal disorders and headache. No clinically meaningful increase of adverse events was observed in prucalopride treated groups as compared to placebo group.
A total of 564 elderly patients (≥ 65 years) with chronic constipation were treated with prucalopride in double-blind studies, with a total exposure of 63 person-years. Most patients in the phase II/III double-blind placebo-controlled studies were younger than 65 years. The incidence of adverse events in the < 65 years old group was 71.2% (1534 out of 2153 patients) in the prucalopride group, and 61.6% (712 out of 1155) in the placebo group. In the group of patients older than 65 years, the incidence of adverse events in the prucalopride group was 58.7% (331 out of 564) and in the placebo group 52.8% (113 out of 214). Similar to the younger age group, the most common adverse events with prucalopride treatment among the elderly (> 65 years) groups were gastrointestinal disorders and headache. No clinically meaningful increase of adverse events was observed in prucalopride treated groups as compared to placebo group.
The following adverse reactions were reported in controlled clinical studies at the recommended dose of 2 mg with frequencies corresponding to:
Very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (> 1/1,000 to < 1/100), rare (> 1/10,000 to < 1/1,000) and very rare (≤ 1/10,000).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. Frequencies are calculated based on the placebo-controlled clinical study data.
Metabolism and nutrition disorders.
Common: decreased appetite.
Nervous system disorders.
Very common: headache. Common: dizziness. Uncommon: tremors, migraine.
Cardiac disorders.
Uncommon: palpitations.
Ear and labyrinth disorders.
Uncommon: vertigo.
Gastrointestinal disorders.
Very common: nausea, diarrhoea, abdominal pain. Common: vomiting, dyspepsia, flatulence, abnormal bowel sounds. Uncommon: rectal haemorrhage.
Renal and urinary disorders.
Common: polyuria.
General disorders and administration site conditions.
Common: fatigue. Uncommon: fever, malaise.
After the first day of treatment, the most common adverse reactions were reported in similar frequencies (incidence less than 1% difference between prucalopride and placebo) during prucalopride therapy as during placebo, with the exception of nausea and diarrhoea that still occurred more frequently during prucalopride therapy, but less pronounced (difference in incidence between prucalopride and placebo between 1 and 3%).
Palpitations were reported in 0.7% of the placebo patients, 1.0% of the 1 mg prucalopride patients, 0.7% of the 2 mg prucalopride patients and 1.9% of the 4 mg prucalopride patients. The majority of patients continued using prucalopride. As with any new symptom, patients should discuss the new onset of palpitations with their physician.
Prucalopride has been shown not to cause rebound phenomena, nor to induce dependency.
A thorough QT study was performed to evaluate the effects of prucalopride on the QT interval at therapeutic (2 mg) and supratherapeutic doses (10 mg) and compared with the effects of placebo and a positive control. This study did not show significant differences between prucalopride and placebo at either dose, based on mean QT measurements and outlier analysis. This confirmed the results of two placebo controlled QT studies. In double blind clinical studies, the incidence of QT-related adverse events and ventricular arrhythmias was low and comparable to placebo.
Suicidal behaviour/ideation.
In the double-blind clinical trials, one patient reported a suicide attempt 7 days after the end of treatment with prucalopride 2 mg once daily; none were reported in patients on placebo. In the open-label trials, two patients reported a suicide attempt and another patient reported suicidal ideation. Completed suicide was reported in two patients, previously treated with prucalopride 2 mg or 4 mg; both discontinued prucalopride for at least one month prior to the event.
Cardiovascular safety analysis.
An evaluation was performed by an independent adjudication committee of all potential major adverse cardiovascular events (MACE) across 28 completed double-blind and open-label clinical studies for prucalopride in adult patients with chronic idiopathic constipation. The standardized incidence rate (IR) per 1000 subject-years for MACE for prucalopride was compared with the IR for placebo. The total exposure in the double-blind studies was 565.2 subject-years in the prucalopride group, 384 subject-years in the placebo group and 2769 subject-years in the double-blind and open-label clinical studies. The IR for MACE was 3.5 (2 subjects out of 3366) in the double-blind prucalopride group, 5.2 (2 subjects out of 2019) in the placebo group, and 3.3 (9 subjects out of 4472) for prucalopride in the combined double-blind and open-label clinical studies. The data do not indicate an increased risk of MACE attributable to prucalopride when compared to placebo.
Observational cardiovascular cohort study.
The overall (CV) safety of prucalopride was assessed in an observational population-based cohort study using European healthcare databases. New users of prucalopride (N=5715) were matched to new users of polyethylene glycol 3350 (PEG) (N=29,372) to determine the standardized incidence rate (IR) and the adjusted incidence rate ratio (IRR) per 1,000 person-years for MACE. In this cohort study, the pooled, standardized IR for MACE was 6.57 (95% CI: 3.90, 10.39) for prucalopride compared to an IR of 10.24 (95% CI: 6.97, 14.13) for PEG and the IRR for MACE was 0.64 (95% CI: 0.36, 1.14). These data do not indicate an increased risk of MACE in patients using prucalopride as compared with patients using PEG for chronic idiopathic constipation.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
In a study in healthy volunteers, treatment with prucalopride was well tolerated when given in an up-titrating scheme up to 20 mg once daily (10 times the recommended therapeutic dose). An overdose may result in symptoms resulting from an exaggeration of the medicinal product's known pharmacodynamic effects and include headache, nausea and diarrhoea. Specific treatment is not available for prucalopride overdose. Should an overdose occur, the patient should be treated symptomatically and supportive measures instituted, as required. Extensive fluid loss by diarrhoea or vomiting may require correction of electrolyte disturbances.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Pharmacotherapeutic group: drugs acting on serotonin receptors, ATC code: A03AE04.
In subjects with chronic constipation, there was a reduction in small bowel transit time, an increase in gastric emptying and more rapid colonic filling. There was an increase in the frequency of bowel motions but no significant effect on colonic transit time.
Clinical trials.
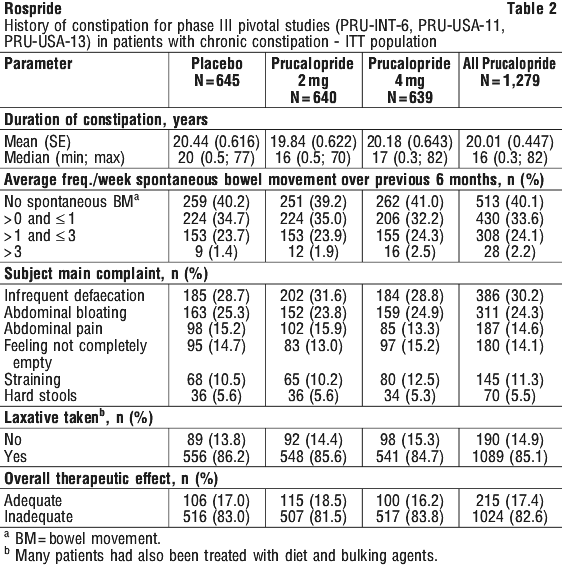
The efficacy of prucalopride was established in three multicentre, randomised, double-blind, 12 week placebo-controlled studies in subjects with chronic constipation (n=1,279 on prucalopride, 1,124 females, 155 males) namely PRU-INT-6, PRU-USA-11 and PRU-USA-13. The studies consisted of 2 phases: a 2-week drug-free run-in phase followed by a randomised, 12-week, double-blind, placebo-controlled treatment phase. The prucalopride doses studied in each of these three studies included 2 mg and 4 mg once daily. The respective mean ages of patients in the three studies were 43.9, 48.3, and 47.9 (range 17-95) years. Patients with secondary causes of constipation including opioid use, endocrine disorders, metabolic disorders and neurologic disorders were excluded from the studies. Table 2 provides a summary of the constipation history (prior to study enrolment) demonstrating that the patients enrolled were chronically constipated. Over 70% of patients had ≤ 1 SBM at baseline and more than 80% indicated that prior therapy was inadequate.
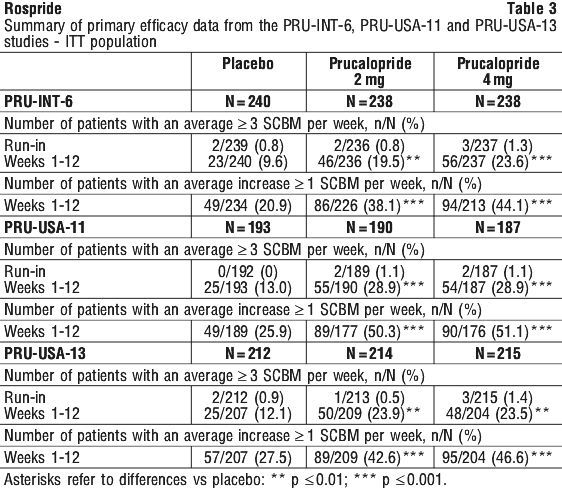
 The primary efficacy endpoint was the proportion (%) of subjects that reached normalisation of bowel movements defined as an average of three or more spontaneous, complete bowel movements (SCBM) per week over the 12-week treatment period. The main secondary efficacy parameter was the proportion of patients with an average increase of ≥ 1 SCBM per week from run-in. A summary of primary efficacy data for individual pivotal studies is provided in Table 3. Both doses were statistically superior (p < 0.001) to placebo at the primary endpoint in each of the three studies, with no incremental benefit of the 4 mg over the 2 mg dose.
The primary efficacy endpoint was the proportion (%) of subjects that reached normalisation of bowel movements defined as an average of three or more spontaneous, complete bowel movements (SCBM) per week over the 12-week treatment period. The main secondary efficacy parameter was the proportion of patients with an average increase of ≥ 1 SCBM per week from run-in. A summary of primary efficacy data for individual pivotal studies is provided in Table 3. Both doses were statistically superior (p < 0.001) to placebo at the primary endpoint in each of the three studies, with no incremental benefit of the 4 mg over the 2 mg dose.
 Results from the analyses of proportion of patients achieving an average of ≥ 3 SCBM per week during weeks 1-4 were similar to that for the primary efficacy endpoint. Both doses were significantly superior (p < 0.001) to placebo in each of the three studies.
Results from the analyses of proportion of patients achieving an average of ≥ 3 SCBM per week during weeks 1-4 were similar to that for the primary efficacy endpoint. Both doses were significantly superior (p < 0.001) to placebo in each of the three studies.
In the pooled 3 pivotal study data analyses, the proportion of patients treated with the recommended dose of 2 mg prucalopride that reached an average of ≥ 3 SCBM per week was 27.8% (week 4) and 23.6% (week 12), versus 10.5% (week 4) and 11.3% (week 12) on placebo. A clinically meaningful improvement of ≥ 1 SCBM per week, the most important secondary efficacy endpoint, was achieved in 48.1% (week 4) and 43.1% (week 12) of patients treated with 2 mg prucalopride versus 23.4% (week 4) and 24.6% (week 12) of placebo patients. Based on the 12 weeks pooled data, for placebo, 1 out of 8 patients responded (i.e. had ≥ 3 SCBM/week). For an average number of 8 patients receiving prucalopride 2 mg daily, one additional patient had ≥ 3 SCBM per week (i.e. NNT=8), indicating that for patients on prucalopride 2 mg, 2 out of 8 patients responded. For an average of 7 patients who received prucalopride 4 mg daily, one additional patient had ≥ 3 SCBM/week compared with the placebo control (i.e. NNT=7). For a clinically meaningful improvement of ≥ 1 SCBM/week, for placebo, 1 out of 4 patients had ≥ 1 SCBM/week. For an average of 5 patients receiving prucalopride 2 mg daily or 4 patients receiving prucalopride 4 mg daily, one additional patient had ≥ 1 SCBM/week, compared with the placebo control (i.e. NNTT=5 for the 2 mg daily, and NNTT=4 for the 4 mg daily).
In all three studies, prucalopride significantly improved the time to first bowel movement when compared with placebo. In addition, for all three studies, treatment with prucalopride resulted in significant improvements in a validated and disease specific set of symptom measures (PAC SYM), including abdominal, stool and rectal symptoms, determined at week 4 and week 12. A significant benefit on a number of quality of life measures, such as degree of satisfaction with treatment and with bowel habits, physical and psychosocial discomfort and worries and concerns, was also observed at both the 4 and 12 week assessment time points.
Other clinical studies.
Study INT-12 was a double-blind, placebo-controlled study that evaluated the efficacy, safety and quality-of-life of prucalopride in 303 elderly patients (≥ 65 years) with chronic constipation. The doses studied were 1 mg, 2 mg and 4 mg. The primary efficacy endpoint was the proportion of patients with an average of ≥ 3 SCBM per week evaluated over the 4-week treatment period. The key secondary efficacy endpoint was the proportion of patients with an average increase of ≥ 1 SCBM per week. Results showed that there was a higher proportion of patients in all 3 prucalopride groups with ≥ 3 SCBM per week compared to placebo, although this observation was not statistically significant. The proportion of patients with an average increase of ≥ 1 SCBM per week was significantly higher for all 3 prucalopride treatment groups when compared to placebo. The results showed that no advantage was gained by increasing the dose beyond 1 mg.
Over 600 elderly subjects were investigated in double-blind placebo-controlled phase II and III studies comparing the 0.5 mg, 1 mg, 2 mg and 4 mg doses of prucalopride with placebo. Results demonstrated that the 1 mg daily dose is the lowest effective dose in achieving the primary endpoint of ≥ 3 SCBM per week and the secondary endpoint of increase ≥ 1 SCBM per week.
Data from open label studies up to 2.6 years offer some evidence for longer-term safety and efficacy; however, no placebo controlled efficacy data for treatments longer than 12 weeks duration are available.
5.2 Pharmacokinetic Properties
Absorption.
Prucalopride is rapidly absorbed; after a single oral dose of 2 mg, Cmax was attained in 2-3 hours. The absolute oral bioavailability is > 90%. Concomitant intake of food does not influence the oral bioavailability of prucalopride.
Distribution.
Prucalopride is extensively distributed and has a steady-state volume of distribution (Vdss) of 567 L. The plasma protein binding of prucalopride is about 30%.
Metabolism.
Metabolism is not the major route of elimination of prucalopride. In vitro, human liver metabolism of prucalopride is very slow and only minor amounts of metabolites are found. In an oral dose study with radio-labelled prucalopride in man, small amounts of eight metabolites were recovered in urine and faeces. The major metabolite (R107504, formed by O-demethylation and oxidation of the resulting alcohol function to a carboxylic acid) accounted for less than 4% of the dose. Unchanged active substance made up about 85% of the total radioactivity in plasma and only R107504 was a minor plasma metabolite.
Excretion.
A large fraction of the active substance is excreted unchanged (about 60% of the administered dose in urine and at least 6% in faeces) via both passive filtration and active renal transporters (P-gp and BCRP). Renal excretion of unchanged prucalopride involves both passive filtration and active secretion. The plasma clearance of prucalopride averages 317 mL/min. Its terminal half-life is about one day. Steady-state is reached within three to four days. On once daily treatment with 2 mg prucalopride, steady-state plasma concentrations fluctuate between trough and peak values of 2.5 and 7 nanogram/mL, respectively. The accumulation ratio after once daily dosing ranged from 1.9 to 2.3. The pharmacokinetics of prucalopride is dose-proportional within and beyond the therapeutic range (tested up to 20 mg). Prucalopride o.d. displays time-independent kinetics during prolonged treatment.
Special populations.
Population pharmacokinetics.
A population pharmacokinetic analysis showed that the apparent total clearance of prucalopride was correlated with creatinine clearance, and that there was no additional effect of age, body weight, sex or race.
Elderly.
After once daily dosing of 1 mg, peak plasma concentrations and AUC of prucalopride in elderly subjects were 26% to 28% higher than in young adults. This effect can be attributed to a diminished renal function in the elderly.
Renal impairment.
Compared to subjects with normal renal function, plasma concentrations of prucalopride after a single 2 mg dose were on average 25% and 51% higher in subjects with mild (ClCR 50-79 mL/min) and moderate (ClCR 25-49 mL/min) renal impairment, respectively. In subjects with severe renal impairment (ClCR ≤ 24 mL/min), plasma concentrations were 2.3 times the levels in healthy subjects (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Hepatic impairment.
Non-renal elimination contributes to about 35% of total elimination. After a single oral dose of 2 mg, Cmax and AUC of prucalopride were on average 10-20% higher in patients with moderate and severe hepatic impairment than in subjects with normal hepatic function.
Paediatric.
After a single oral dose of 0.03 mg/kg in paediatric patients aged between 4 and 12 years, Cmax of prucalopride was comparable to the Cmax in adults after a single 2 mg dose, while unbound area under the curve (AUC) was 30-40% lower than after 2 mg in adults. Unbound exposure was similar over the whole age-range (4-12 years). The average terminal half-life in the paediatric subjects was about 19 hours (range 11.6 to 26.8 hours) (see Section 4.2 Dose and Method of Administration). Prucalopride is not recommended in children or adolescents (see Section 4.4 Special Warnings and Precautions for Use, Paediatric use).
5.3 Preclinical Safety Data
Genotoxicity.
The standard bacterial reverse mutation test had a weak positive result in one of the five strains at high concentrations (≥ 0.5 mg/plate). All subsequent in vivo tests on gene mutation, chromosomal damage, unscheduled DNA repair and DNA adduct induction showed negative results, which demonstrated that prucalopride did not have genotoxic potential.
Carcinogenicity.
Carcinogenicity studies were conducted with oral prucalopride doses up to 80 mg/kg/day in mice, and 40 (female) and 80 (male) mg/kg/day in rats, for two years. In mice, the incidence of mammary gland adenocarcinomas was increased at 80 mg/kg/day (200 times clinical exposure at the MRHD, based on AUC); the no-effect dose was 20 mg/kg/day (27 times clinical exposure at the MRHD, based on AUC). In rats, the high doses were associated with increased incidences of benign adrenal phaeochromocytomas, pituitary adenomas, pancreatic adenomas, hepatocellular adenomas (mid and high doses) and thyroid follicular tumours (45 times clinical exposure at MRHD, based on AUC); the no adverse effect dose was 5 mg/kg/day (7 times clinical exposure at the MRHD, based on AUC). Mechanistic studies showed that hyperprolactinaemia resulted from D2 antagonism at high prucalopride concentrations likely caused the mammary, pituitary, pancreatic and adrenal tumours in both mice and rats. Prucalopride and its rat specific metabolism at high doses had hepatic enzyme induction potential that led to the liver and thyroid tumours in rats. Since no increase of plasma prolactin levels was observed in clinical studies and human prucalopride metabolism was very different from that of the rat, these tumour findings were considered to have minimum clinical relevance.6 Pharmaceutical Particulars
6.1 List of Excipients
Prucalopride tablets contain the following inactive ingredients:
Tablet core.
Lactose monohydrate, microcrystalline cellulose, magnesium stearate, colloidal anhydrous silica.
Coating.
Hypromellose, titanium dioxide, macrogol 400, polysorbate 80, iron oxide red (2 mg tablet only).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Prucalopride tablets should be kept out of reach of children. Store below 25°C. Store in the original blister in order to protect from moisture.
6.5 Nature and Contents of Container
Both strengths of prucalopride film-coated tablets are available in Alu - OPA/Alu/PVC or Alu - PVC/PE.EVOH.PE/PCTFE perforated unit dose blisters containing 28 film-coated tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Prucalopride succinate is a white to almost white powder.
Chemical structure.
The chemical name for prucalopride is 4-amino-5-chloro-2,3-dihydro-N-[1-(3-methoxypropyl)-4- piperidinyl]-7-benzofurancarboxamide butanedioate (1:1). Prucalopride has the following chemical structure:
 C18H26ClN3O3.C4H6O4. Molecular weight: 485.96.
C18H26ClN3O3.C4H6O4. Molecular weight: 485.96.
CAS number.
179474-85-2.
Solubility.
In organic media, prucalopride succinate is soluble in N,N-dimethylformamide, sulfinylbismethane and N, N-dimethylacetamide and sparingly soluble in methanol. In aqueous media, prucalopride succinate is freely soluble in acidic aqueous media. However, this solubility decreases with increasing pH.
Dissociation constant.
The pKa for the piperidine moiety of prucalopride succinate is 8.5, determined at 20°C by potentiometric titration of an aqueous solution of prucalopride succinate. The pKa for the amino moiety of prucalopride succinate is less than 3, determined at 20°C by spectrometric measurements of solutions of prucalopride succinate in water at different pH-values.
Partition coefficient.
The partition coefficients are determined at 20°C between n-octanol and aqueous buffered solutions. The partition coefficient P is defined as the ratio of the equilibrium concentrations of a single molecular species in a two-phase system of n-octanol and an aqueous buffered solution. The results for partition coefficient P are shown in Table 4.

7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes
